










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAgronomía Mesoamericana
On-line version ISSN 2215-3608Print version ISSN 1659-1321
Agron. Mesoam vol.22 n.2 San Pedro Dec. 2011
Análisis y comentarios
Uso correcto del análisis clúster en la caracterización de germoplasma vegetal
Carlos Alberto Núñez-Colín2*, Diana Escobedo-López2
*Dirección de correspondencia:
Resumen
Uso correcto del análisis clúster en la caracterización de germoplasma vegetal. El presente trabajo tuvo como objetivo hacer una recopilación de información básica para poder realizar de forma apropiada el análisis clúster. Este método multivariado permite hacer agrupaciones y es muy utilizado en estudios de caracterización de recursos genéticos. Se mencionan los tipos de datos a utilizar, los índices de similitud y disimilitud, los métodos de aglomeración y la forma correcta de establecer el número de grupos en un dendrograma.
Palabras clave: Métodos estadísticos multivariados, índices de similitud y disimilitud, partición de dendrogramas.
Abstract
Proper use of the cluster analysis in plant germplasm characterization. The present essay aims to make a recompilation of basic information to properly perform the cluster analysis. This multivariate method allows making groups and it is widely used in studies of genetic resources characterization. It is mentioned topics like the data types to use, similarity and dissimilarity indices, agglomerative methods, and the correct way to establish the number of groups into a dendrogram.
Key words: Multivariate statistical methods, similarity and dissimilarity indices, dendrogram partitioning.
Introducción
Actualmente en México, el estudio de los recursos fitogenéticos es una línea de investigación que ha adquirido gran importancia, debido a que el país es considerado como mega diverso (Ramamoorthy et al. 1993), y en donde se encuentran muchas especies potenciales que no han sido debidamente estudiadas. En este sentido, las herramientas estadísticas utilizadas, sobretodo en la caracterización de germoplasma, son tópicos en los que la mayoría de los investigadores, sobretodo los que han adoptado este enfoque, no han explorado ni estudiado a fondo sus bases.
En artículos publicados recientemente, se desconoce el buen uso de estas herramientas, además de exitir errores en la interpretación de los resultados; siendo el análisis de agrupación, más conocido como análisis clúster, uno de los métodos más comunes para este tipo de estudios.
El presente trabajo tuvo como objetivo hacer una recopilación de información básica para poder realizar de forma apropiada el análisis clúster.
Unidad básica de caracterización
Como primer punto para las futuras explicaciones en este ensayo sobre el análisis clúster, se definirá el concepto de Unidad Básica de Caracterización (UBC) que es definida como la unidad básica del objeto (planta, animal o entidad) que se va ha describir y que a su vez será empleada para lograr el objetivo de la caracterización; dependiendo del propósito del estudio de caracterización, la UBC puede ser un individuo, una población silvestre, una línea o un híbrido. (González-Andrés 2001). Anteriormente a las UBC se les generalizaba con el nombre de Unidad Taxonómica Operativa (OTU, por sus siglas en inglés) (Sokal y Sneath 1963); sin embargo, como no todos los estudios de caracterización están enfocados a la taxonomía, el término correcto debería ser UBC.
Definiciones básicas en los Análisis Clúster
El análisis clúster es un método matemático que está incluido en lo que hoy se llama estadística multivariada o estadística multivariante; este método es principalmente utilizado para la formación de grupos de UBC’s con características similares a partir de las similitudes o disimilitudes que se presentan entre pares de estas UBC’s en n características evaluadas (Johnson 1998). Este tipo de análisis está compuesto por dos métodos interrelacionados e igualmente importantes. El primero es el cálculo de los índices de similitud o de disimilitud entre pares de UBC’s, del cual existe un sin número de referencias en la literatura; no obstante, estos índices deben ser aplicados de acuerdo a la naturaleza de los datos y al objetivo de la caracterización. Y el segundo es la aplicación del método de aglomeración adecuado, donde también existe mucha literatura al respecto, que permite a partir de los índices de similitud o disimilitud generar las gráficas de árbol o dendrogramas que son representaciones gráficas donde el investigador puede tener de una manera resumida el parecido que presentan los grupos de UBC’s. El método de aglomeración a utilizar principalmente está definido por el objetivo de la caracterización.
Los métodos, cálculo de los índices de similitud y la aplicación del método de aglomeración, englobados en el análisis clúster, a pesar de que presentan unas sólidas bases matemáticas no son tan restrictivos respecto a sus bases estadísticas (Johnson 1998). Por lo que el principal problema que presenta este análisis es la elección del índice de similitud o disimilitud y del método de aglomeración más apropiados; la elección de la mejor combinación de los métodos del análisis clúster depende principalmente de la naturaleza de los datos (si son datos doble estado, multi-estado con o sin secuencia lógica, cuantitativos, genéticos o secuencias de ADN/proteínas) y el objetivo de la caracterización. Caracterizar es un método no es un objetivo.
Naturaleza de los datos
Dentro de la taxonomía numérica definida por Sneath y Sokal (1962) se definió parte de la siguiente clasificación para la naturaleza de los datos para caracterizaciones. Con base en esta clasificación se diseñaron, o se rescataron en algunos casos, los índices de similitud o disimilitud que se utilizan en la taxonomía numérica. Asimismo, los datos genéticos y los de secuencias de ADN y proteínas fueron definidos por sus características particulares con base en la genética de poblaciones, a la genética moderna, y a la filogenia moderna, así como también estas disciplinas definen a sus índices de disimilitud (Cavalli-Sforza y Edwards
1967, Jukes y Cantor 1969, Nei 1972).
Datos doble estado
Los datos doble estado son obtenidos cuando sólo se presentan en la característica evaluada dos posibilidades de respuesta; por ejemplo, ausencia o presencia. Estas generalmente son codificadas como 0 para las negativas y 1 para las positivas; por ejemplo, 0 para ausencia y 1 para presencia. Estos datos al tener esta respuesta presentan una distribución estadística binomial y el cálculo de sus estadísticos básicos debe realizarse con las fórmulas diseñadas para esta distribución.
Ejemplos de estos caracteres son el uso de claves dicotómicas de taxonomía, la codificación de presencias/ausencias de huellas moleculares, características morfológicas que sólo presentan dos estados, etcétera.
Datos multi-estado
Los datos multi-estado son datos cualitativos, y a diferencia de los de doble estado, presentan más de dos posibilidades de respuesta; pero una complicación más, y es que estos pueden ser con secuencia lógica o sin esta, es decir, este tipo de datos engloba a los estadísticos nominales (sin secuencia lógica) y a los ordinales (con secuencia lógica), cada uno de ellos con características propias y métodos diseñados ex professo para cada uno.
Los datos nominales son aquellos con más de dos posibilidades de respuesta pero que no llevan un orden lógico. Por ejemplo, a los colores se les puede poner un número, es decir, rojo = 1, amarillo = 2, azul = 3, etc; sin embargo, no se puede concluir que rojo es menor que azul, son diferentes pero no en orden o secuencia lógica.
Por otro lado, los datos ordinales, al tener más de dos posibilidades de respuesta, pueden ser ordenados con una secuencia lógica. Por ejemplo, tamaño de hoja que puede ser chica = 1, mediana = 2, grande = 3. En este caso si se puede decir que grande es más que chica o que la mediana aunque no de una manera cuantitativa. Estos datos cumplen generalmente con las distribuciones polinomiales u otras distribuciones derivadas de variables discretas. No obstante, datos ordinales y nominales son estadísticamente diferentes.
Ejemplos del uso de estos son los caracteres morfológicos diferenciales a los que no se les mide cuantitativamente sus diferencias; por ejemplo, tipo de copa de árboles, densidad de pubescencia, colores de frutos, todas las escalas de medición por daños, etc.
Datos cuantitativos
Los datos cuantitativos son los que pueden contarse y que son continuos (presentan cualquier valor real); generalmente con una distribución normal e incluso algunos datos discretos pueden ser utilizados con un buen muestreo pero mediante estadísticos de tendencia central para obtener normalidad. Ejemplos del uso de estos caracteres son altura de planta, peso de fruto y de semilla, materia seca, color en escalas Hue, Croma y L; además de número de hojas, de frutos y de flores, entre otras (si el muestreo es adecuado). Estos datos en la actualidad son los que más se utilizan en estudios de caracterización morfológica aunque más bien debería ser llamada caracterización morfométrica.
Datos genéticos
Los datos genéticos son aquellos en los cuales el investigador puede conocer características que cumplan con la genética mendeliana y de poblaciones, es decir, donde se conocen el número de loci que se está evaluando y el número de alelos por locus.
Generalmente los datos que se requieren para caracterización genética, son frecuencias alélicas y génicas por población (Cavalli-Sforza y Edwards 1967, Nei 1972, Nei 1978); esta población debe ser lo suficientemente grande para poder obtener los datos (Wright 1978). No se pueden obtener datos genéticos con poblaciones pequeñas y menos aún con individuos únicos porque se pueden evaluar alelos atípicos en la muestra, los cuales pueden afectar las conclusiones de la investigación (Falconer y Mackay 2001).
Secuencias de ADN y proteínas
Actualmente, con la tecnología moderna, se han desarrollado herramientas que permiten a los investigadores obtener secuencias de nucleótidos de ADN, principalmente de genes conservados, así como la secuencia de aminoácidos de las proteínas. De ambos tipos de secuencias se han estudiado sus características particulares por diversos autores (Fuerst et al. 1977, Chakraborty et al. 1978, 1980, Nei y Gojobori 1986, Nei y Miller 1990, Tamura y Nei 1993), lo que ha permitido que pueden detectarse los cambios que han sufrido a través del tiempo así como el parecido filogenético que presentan dos UBC’s en estas secuencias.
Por las características especiales de estos datos, se han desarrollado métodos apropiados para analizarlos (Kumar et al. 2008). Estos generalmente, son usados para estudios filogenéticos aunque en algunos casos han servido para descifrar aspectos taxonómicos, además de los puramente filogenéticos, con géneros complejos como en el caso de Crataegus (Lo et al. 2007).
Índices de similitud y disimilitud
Una vez que se tienen detectados el tipo de datos que van a utilizarse en el estudio de caracterización, se procede a seleccionar el mejor índice de similitud o de disimilitud apropiado. La primera recomendación es no hacer combinación de datos; esto es debido a que, como ya se mencionó anteriormente, cada tipo de datos presenta características propias que no comparte con los de otra naturaleza.
Índices de similitud
Los primeros índices que se abordarán serán los de similitud, estos están desarrollados, principalmente, para datos doble estado. Aunque algunos de estos índices pueden ser utilizados también para analizar caracteres multi-estado sin secuencia lógica (nominales). Todos ellos basados en los mismos principios que se describen a continuación:
Suponga a dos UBC’s a las que se les ha obtenido datos doble estado, las combinaciones de comparación posibles se muestran en el Cuadro 1.

Donde a es que los dos individuos tengan presente la misma característica; b es que el primero la presente y el segundo no; c que el segundo presente la característica y el primero no, y d que ambos carezcan de esta característica. A partir de esto calculamos m que es la suma entre a y d (m = a + d), es decir, la suma de las concordancias, tanto de presencia como de ausencia; u que es la suma de b y c (u = b + c), es decir, la suma de las discordancias, y n que es el número total de características evaluadas, es decir, la sumas de las concordancias y las discordancias (n = m + u). A partir de esta codificación tenemos las siguientes fórmulas (Cuadro 2). Estas codificaciones son las que se utilizan en la actualidad a nivel mundial en los artículos relacionados con la teoría y el desarrollo de estos índices.
Hay que tener claro que cada uno de estos índices fueron desarrollados para fines específicos; por ejemplo, Simple matching (Sokal y Sneath 1963) fue desarrollada para analizar características evaluadas a partir de claves taxonómicas dicotómicas, y en donde un ejemplar podía o no tener ciertas características y en raros casos se presentaban más de dos posibilidades de respuesta; esta es la razón por la cual este índice si permite datos multi-estado sin secuencia lógica.
Sin embargo, si lo que se va a analizar son matrices binomiales de presencia/ausencia de bandas de huellas de ADN o de proteínas, obtenidas por electroforesis, el índice más adecuado de utilizar es la fórmula desarrollada por Dice (1945), que, aunque ésta fue desarrollada originalmente con fines de comparación de nichos ecológicos, Nei y Li (1979) demostraron, mediante desarrollos matemáticos, que era el índice más congruente para la clasificación mediante estas características debido principalmente a que una concordancia por doble ausencia de una banda puede no deberse a la misma razón, es decir, no es una concordancia. Por este y otros muchos ejemplos, el investigador tiene que tener nociones de dónde se obtuvo y para qué se utiliza el índice de similitud que va a usar; esta recomendación es para no tener problemas con la interpretación de sus resultados o, lo más común, hacer conclusiones que no son ciertas y que no aportan nada al conocimiento científico mundial.
Distancias métricas (morfológicas o taxonómicas)
Los índices de disimilitud, mejor conocidos como distancias, están basadas en planos euclidianos n–dimensionales (Lindgren 1968).
Las distancias están desarrolladas para aplicarse a datos multi-estado con secuencia lógica y a los datos cuantitativos; no obstante, hay distintas distancias para cada tipo de datos por lo que no es recomendable aplicarlo a combinaciones de ellos. La distancia más básica es la euclidiana que se basa en el teorema de Pitágoras; esta distancia es la raíz cuadrada de la suma de los cuadrados de cada una de las diferencias (cada uno puede ser considerado un cateto) para así obtener la distancia (que sería la hipotenusa) entre esos dos puntos. Para ejemplificarlo se hará el modelo más simple con dos variables (Figura 1), pero este modelo es válido para n catetos en n dimensiones.
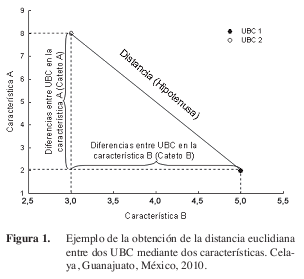
A partir de esta distancia se han hecho modificaciones para fines específicos de los cuales se han desarrollado las siguientes fórmulas (Cuadro 3).
La distancia más utilizada en estudios morfométricos (variables cuantitativas) es la distancia euclidiana media, o también llamada distancia taxonómica media, que tiene como variante dividir la distancia entre el número de características antes de obtener la raíz cuadrada, esto con el fin de tener escalas comparables, porque cuando se comparan dos distancias en investigaciones diferentes, estas distancias pueden comparase aún cuando no se tengan el mismo número de características evaluadas.
Otra variante importante es la distancia χ2, que está desarrollada para datos multi-estado con secuencia lógica (cualitativos ordinales) o, en su defecto, rangos de valores de datos cuantitativos, aunque se puede usar para datos cuantitativos, se debe de tener cuidado en utilizar escalas equiparables en todas las variables que se pretenda incluir en la investigación (Núñez-Colín et al. 2004), por lo que su uso se debe de hacer sólo en situaciones particulares.
Existen distancias que son poco utilizadas en análisis clúster; sin embargo, son empleadas en otro tipo de análisis como lo es el discriminante canónico, este es el caso de la distancia Mahalanobis que es una modificación de la distancia euclidiana vista desde un punto de vista del álgebra de matrices, donde, además de las diferencias de los caracteres entre UBC’s, utiliza la matriz de varianzas y covarianzas entre ambas UBC’s en su cálculo, esta distancia cuanta con pruebas de hipótesis que permiten conocer si dos UBC’s son o no diferentes.
Distancias genéticas
A diferencia de las distancias métricas, las genéticas son utilizadas para conocer la disimilitud de caracteres genéticos. Por lo tanto, se necesita de caracteres codificados de acuerdo a la genética de poblaciones, es decir, número de loci a analizar así como número de alelos para cada locus; esto puede obtenerse a partir de caracteres morfológicos de herencia mendeliana o de marcadores moleculares aplicados a poblaciones (nunca aplicados únicamente a individuos) donde se puedan detectar los parámetros de frecuencias alélicas y génicas. Las distancias genéticas más utilizadas se encuentran resumidas a continuación (Cuadro 4).
En el caso de las distancias genéticas, la más utilizada es la fórmula original de Nei que, aunque puede no ser imparcial por el orden en el que se meten los datos, es la que mejores resultados ha dado en investigaciones filogenéticas. Sin embargo, existen fórmulas muy bien estructuradas en bases genéticas y filogenéticas como las desarrolladas por Cavalli-Sforza y Edwards (1967) pero que por su complejidad no son muy utilizadas, aunque también son válidas para estudios de este tipo.
Todas las fórmulas de distancias genéticas fueron obtenidas para datos de frecuencias alélicas, sobre todo de las génicas, y que, aunque pueden ser utilizadas por otro tipo de datos, da como resultado una interpretación totalmente incongruente. Un caso común de esto es cuando a datos binarios de huellas genéticas se les quiere aplicar alguna de estas fórmulas dando como resultado que, aunque se tenga un dato de las distancias, la interpretación del parecido entre ellas no es congruente en comparación a si se aplicara un índice de similitud que sería lo correcto.
Distancias genéticas para secuenciación
En el caso de las distancias genéticas para secuenciación, también existen un sin número de fórmulas en la literatura, todas ellas basadas en los patrones de sustitución de bases para el caso de ADN (transiciones y transversiones) y de aminoácidos para el caso de proteínas; todas ellas cumpliendo con las teorías desarrolladas en la genética moderna sobre la mutación del ADN y de las proteínas (Tamura et al. 2007).
Métodos de aglomeración
En cuanto a los métodos de aglomeración o de agrupamiento también se tienen diferencias entre ellos porque existen métodos jerárquicos y no jerárquicos, siendo los más comúnmente utilizados los jerárquicos, aunque en la actualidad se han hecho modificaciones a algunos de estos métodos para tener representaciones no jerárquicas, estos tienen una base matemática fuerte, pero no desde el punto de vista biológico.
El principio de todos los métodos es unir las UBC’s con el máximo parecido en la matriz de distancias o en la matriz de índices de similitud, a este grupo de dos UBC’s se le llama nudo; de aquí, se rehace la matriz de similitud o disimilitud para que el nudo tenga un solo valor con respecto a las demás UBC’s, que es donde está la diferencia entre los distintos métodos, después de este paso se puede formar otro nudo entre UBC’s diferentes o unir otra UBC diferente a un nudo previamente formado; esta operación de rehacer la matriz se repite hasta quedar una sola agrupación (conocida como raíz del dendrograma); de manera que entre más grande sea la distancia o más pequeño sea en índice de similitud entre agrupaciones, más diferentes son.
Los principales métodos jerárquicos y los que poseen modificaciones para su representación no jerárquica se resumen en el Cuadro 5.
El principal problema de los métodos no jerárquicos es no tener un centro fijo (raíz), por lo que el investigador tiene que poseer un amplio conocimiento sobre la especie y el método a utilizar para fijar la raíz correcta del árbol, por lo que es recomendable para investigadores con poca experiencia utilizar preferentemente métodos jerárquicos.
El método más utilizado es el UPGMA, donde el recálculo de la matriz de distancias o de índices de similitud se hace promediando los valores de las distancias o de los índices de similitud de las UBC’s del nudo o grupo con el de las otras UBC’s que es lo matemáticamente más lógico para el recálculo de la matriz.
Un caso especial es el método de Ward (Ward, 1963), el cual sólo es posible para el caso de calcularse a partir de distancias de datos cuantitativos; este método es una alternativa que reduce la presencia de individuos atípicos que se denominan en inglés outliers (individuos raros sin un grupo definido, en muchas ocasiones dado por una única variable con un dato atípico), esto es posible debido a que este método antes de calcular la distancia aplica un análisis en componentes principales y calcula la distancia con los valores de la proyección de los vectores propios de las UBC’s como si fueran las variables originales y recalcula la matriz mediante el método UPGMA. No obstante, se debe tener extrema precaución si al emplear este método se le trata de dar una interpretación taxonómica debido a que se pueden cometer errores serios de interpretación porque este método es más recomendable cuando el objetivo del trabajo es conocer la variabilidad o diversidad existente en UBC’s de una especie o género específico.
Por otro lado, el método Neigthbor joining (Saitou y Nei 1987) debe ser utilizado sólo para estudios filogenéticos y sólo es confiable cuando se ha calculado a partir de distancias genéticas o evolutivas (de frecuencias génicas o datos de secuenciación), y no es recomendable para estudios de caracterización, por lo que se debe de tener cuidado con su uso; actualmente, se tienen programas computacionales que pueden “calcular” este tipo de árboles a partir de índices de similitud o de distancias métricas e incluso existen estudios de comparación de métodos de aglomeración incluyendo este. Sin embargo, la interpretación puede tener muchas complicaciones porque si el árbol de mínima evolución que se obtiene no proviene de datos evolutivos, puede dar una congruencia esperada por el investigador sin que esta sea totalmente correcta, es decir, que se están llegando a conclusiones que al investigador que lo realizó le parecen correctas, o mejor que con otros métodos, pero que si alguien más lo realiza adecuadamente, con datos evolutivos, pueden no ser las mismas. En este sentido, en filogenia también existen otros métodos como máxima parsimonia y máxima verosimilitud, entre otros pero que sólo deben ser empleados con distancias genéticas, principalmente de secuenciación.
Formación de grupos
Otro tópico interesante dentro de los análisis clúster, y donde más errores se cometen, es la definición de la altura de corte de los árboles para definir el número correcto de grupos, para lo cual no existen estadísticos exactos sólo algunas pruebas pseudoestadísticas como la t2 de Hotelling (1951) y el criterio cúbico de agrupación (Cubic Clustering Criterion, CCC) obtenido por Serle (1983). Sin embargo, estas pruebas son difíciles de calcular y no siempre fáciles de interpretar. Por lo que el investigador debe saber cómo cortar su gráfica de árbol o dendrograma.
En este sentido, lo más sencillo es poner una línea recta en alguna parte de dendrograma y contabilizar el número de grupos que se obtienen e ir corroborando su parecido dentro de grupos y entre ellos con otros métodos como el análisis discriminante canónico; pero lo que por ningún motivo debe hacerse es hacer las agrupaciones sólo con el orden en el que se encuentran en el dendrograma porque eso puede variar de acuerdo al programa de cómputo que se utilice y un error muy frecuente es tratar de agrupar individuos atípicos en un mismo grupo, sólo porque aparecen “juntos” en el dendrograma pero con una distancia entre ellos mayor a la de otros grupos.
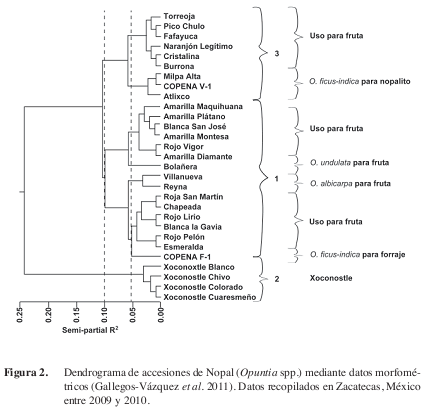
Por ejemplo, Gallegos-Vázquez et al. (2011) utilizando el Método de Ward (Ward 1963) obtuvieron los siguientes resultados (Figura 2) donde se pueden contabilizar tres grupos bien formados. Sin embargo, con una línea de corte más estricta se pueden formar ocho diferentes grupos (que realmente serían subgrupos), donde incluso algunos de ellos son individuos atípicos (en este ejemplo en una se trata de una especie diferente) por lo que ambas alturas de corte son válidas aunque por el concepto de parsimonia (la naturaleza tiende a la sencillez) es siempre recomendable en un primer acercamiento, establecer tres grupos y dependiendo de la interpretación del mismo pueden hacerse más.
Por otro lado, muchas veces quedan individuos atípicos desde el primer corte como lo es en el caso de los marcadores moleculares donde por ejemplo Pecina-Quintero et al. (2011) establecieron dos grupos bien definidos y varios grupos atípicos (Figura 3), la mayoría individuos atípicos, pero no se trató de unir a los grupos mayoritarios porque de esta manera se pierde la interpretación del dendrograma. En este sentido, al establecer una línea de corte más estricta se obtienen más individuos atípicos fuera de los subgrupos.
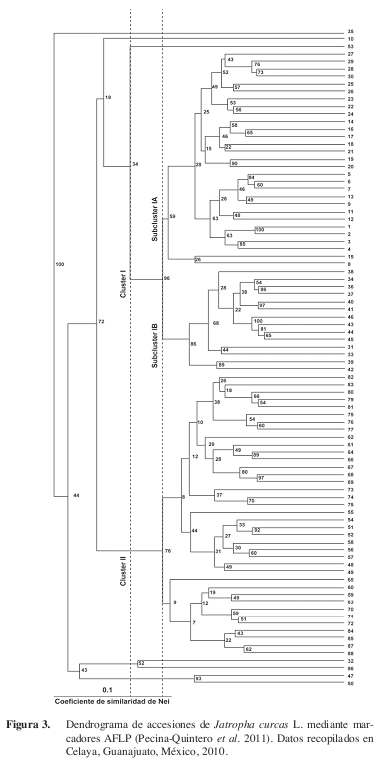
Por lo que lo más difícil de la interpretación del dendrograma es responder a la pregunta ¿Cuántos grupos se deben formar? Por lo que el investigador debe tener una idea clara de, primero, saber qué índice de similitud o disimilitud debe utilizar en cada caso y que método de aglomeración es el adecuado y, segundo, hacer una interpretación válida de su dendrograma: saber cuántos grupos se están formando.
Comentarios finales
Hay que tener nociones de donde salieron los índices de similitud o disimilitud para tener certeza del que se pretenda usar dependiendo del tipo de datos y el objetivo de la caracterización.
El método de aglomeración se debe emplear con base en el tipo de datos y el objetivo de la caracterización.
Las líneas de corte se emplean para definir el número de grupos o hacer pruebas de partición para tener noción de cuantos grupos pueden ser; sin embargo, las pruebas de partición son pseudoestadísticos por lo que no son exactos, sólo ayudan al investigador a darse una idea de cuantos grupos formar por lo que la mejor definición de cuantos grupos es por la experiencia del investigador en estudios similares.
Agradecimientos
Al Dr. Alfredo Josué Gámez Vázquez y al Dr. José Luis Anaya López por sus atinados comentarios de este manuscrito.
Literatura citada
Balakrishnan, V; Sanghvi, LD. 1968. Distance between populations on the basis of attribute data. Biometrics 24 (4): 859-865. [ Links ]
Chakraborty, R; Fuerst, PA; Nei, M. 1978. Statistical studies on protein polymorphism in natural populations. II. Gene differentiation between populations. Genetics 88: 367-390. [ Links ]
Cavalli-Sforza, LL; Edwards, WF. 1967. Phylogenetic analysis models and estimation procedures. Evolution 21: 550-570. [ Links ]
Dice, L. R. 1945. Measure of the amount of ecologic associations between species. Ecology 26: 277-302. [ Links ]
Edwards, WF; Cavalli-Sforza, LL. 1965. A method for cluster analysis. Biometrics 21: 362-375. [ Links ]
Falconer, DS; Mackay, TFC. 2001. Introducción a la genética cuantitativa. Traducido por López-Fanjul de Argüelles, C. ACRIBA, Zaragoza, España. 500 p. [ Links ]
Fuerst, PA; Chakraborty, R; Nei, M. 1977. Statistical studies on protein polymorphism in natural populations. I. Distribution of Single Locus Heterozygocity. Genetics 86:455-483. [ Links ]
Gallegos-Vázquez, C; Barrientos-Priego, AF; Reyes-Agüero, JA; Núñez-Colín, CA; Mondragón-Jacobo, C. 2011. Clusters of commercial cultivars of cactus pear and xoconostle using UPOV morphological traits. Journal of Professional Association for Cactus Development 13:10-23. [ Links ]
González-Andrés, F. 2001 Caracterización morfológica. In González-Andrés, F; Pita Villamil, J. M. eds. Conservación y Caracterización de Recursos Filogenéticos. Publicaciones Instituto Nacional de Educación Agrícola, Valladolid, España. p. 199-217. [ Links ]
Gower, JC. 1967. A comparison of some methods of cluster analysis. Biometrics 23:623-637. [ Links ]
Hamann, U. 1961. Merkmalbestand und Verwandtschaftsbeziehungen der Farinosae. Ein Beitragzum System der Monokotyledonen. Willdenowia 2:639-768. [ Links ]
Hillis, D. M. 1984. Misuse and modification of Nei’s genetic distance. Systems Zoology 33:238-240. [ Links ]
Hotelling, H. 1951. A Generalized t test and measure of Multivariate Dispersion. In Neyman, J. ed. Proceedings of the Second Berkeley Symposium on Mathematical Statistics. University of California Press, Berkeley, USA. p. 23-41. [ Links ]
Jaccard, P. 1908. Nouvelles recherches sur la distribution florale. Bulletin de la Société Vaudoise des Sciences Naturelles 44:223-270.
[ Links ]
Johnson, DE. 1998. Métodos multivariados aplicados al análisis de datos. Traducido por H. Pérez Castellanos. International Thomson Editores, Ciudad de México, México. 566 p. [ Links ]
Jukes, TH; Cantor, CR. 1969. Evolution in protein molecules. In Munro, H. N. (ed.) Mammalian protein metabolism. Academic Press, New York, USA. p. 21-123. [ Links ]
Kulczynski, S. 1927. Die Pflanzenassoziationen der Pienienen. Bull. Intern. Acad. Pol. Sci. Lett. Cl. Sci. Math. Nat., B (Sci. Nat.) (Suppl. 2): 57-203. [ Links ]
Kumar, S; Nei, M; Dudley, J; Tamura, K. 2008. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings in Bioinformatics 9(4):299-306. [ Links ]
Lindgren, BW. 1968. Statistical Theory. Segunda edición McMillanCompany. New York, USA. 521 p. [ Links ]
Lo, E. YY; Stefanovic, S; Dickinson, TA. 2007. Molecular reappraisal of relationships between Crataegus and Mespilus (Rosaceae, Pyreae) – Two genera or one? Systematic Botany 32:596-616. [ Links ]
McQuitty, LL. 1966. Similarity analysis by reciprocal pairs for discrete and continuous data. Educational and Psychological Measurement 26:825-831. [ Links ]
Nei, M. 1972. Genetic distance between populations. The American Naturalist 106 (949):283-292. [ Links ]
Nei, M. 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583-590. [ Links ]
Nei, M; Gojobori, T. 1986. Simple method for estimating the number of synonymous and nonsynonymous nucleotide substitution. Molecular Biology and Evolution 3(5):418-426. [ Links ]
Nei, M; Li, WH. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Science (USA) 76:5269-5273. [ Links ]
Nei, M; Miller, JC. 1990. A Simple Method for Estimating Average Number of Nucleotide Substitutions within and Between Populations from Restriction Data. Genetics 125:873-879. [ Links ]
Núñez-Colín, CA; Rodríguez-Pérez, JE; Nieto-Ángel, R; Barrientos-Priego, AF. 2004. Construcción de dendrogramas de taxonomía numérica mediante el coeficiente de distancia χ2: una revisión. Revista Chapingo Serie Horticultura 10(2):229-237. [ Links ]
Ochiai, A. 1957. Zoogeographic studies on the soleoid fishes found in Japan and its neighbouring regions. Bull. Jap. Soc. Sci. Fish. 22:526-530. [ Links ]
Pecina-Quintero, V; Anay-López, JL; Zamarripa-Colmenero, A; Montes-García, N; Núñez-Colín, CA; SolísBonilla, JL; Aguilar-Rangel, MR; Gill-Langarica, H. R; Mejía-Bustamante, DJ. 2011. Molecular Characterisation of Jatropha curcas L. genetic resources from Chiapas, Mexico through AFLP markers. Biomass and Bioenergy 35(5):1897-1905. [ Links ]
Ramamoorthy TP; Bye R, Lot A, Fa J. (eds). 1993. Biological Diversity of Mexico. Origins and Distribution. Oxford University Press, New York, USA. 812 p. [ Links ]
Rogers, DG; Tanimoto, TT. 1960. A computer program for classifying plants. Science 132:1115-1118. [ Links ]
Rogers, JS. 1972. Measures of genetics similarity and genetics distance. Studies in Gentics VII. University of Texas Publications 7213:145-153. [ Links ]
Russel, PF; Rao, TR. 1940. On habitat and association of species of Anopheline larvae in south-eastern Madras. J. Malar. Inst. India 3:153-178. [ Links ]
Saitou, N; Nei, M. 1987. Neighboor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4:406-425. [ Links ]
Serle, WS. 1983. Cubic clustering criterion. SAS Technical Report A-108. SAS Institute, Cary, USA. 52 p. [ Links ]
Sneath, PHA; Sokal, RR. 1962. Numerical taxonomy. Nature London 193(4818):855-860. [ Links ]
Sokal, RR; Michener, CD. 1958. A statistical method for evaluating systematic relationships. University of Kansas Science Bulletin 38:1409-1438. [ Links ]
Sokal, RR; Sneath, PHA. 1963. Principles of numerical taxonomy. H. Freeman & Company, San Francisco, USA. 359 p. [ Links ]
Sorensen, T. 1948. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content and its application to analyses of the vegetation on danish commons. Biologiske Skrifter 5:1-34. [ Links ]
Swofford, DL; Olsen GJ. 1990. Phylogeny reconstruction. In Hillis, D. M; Moritz, C. (eds.) Molecular systems. Sinauer Association, Sunderland, USA. p. 411-501. [ Links ]
Tamura, K; Nei, M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10(3):512-526. [ Links ]
Tamura, K; Dudley, J; Nei, M; Kumar, S. 2007. MEGA4: Molecular evolutionary genetics analysis (MEGA) Software Version 4.0. Molecular Biology and Evolution 24(8):1596-1599. [ Links ]
Ward, JH. Jr. 1963. Hierarchical grouping to optimize an objective function. Journal of the American Statistical Association 58: 236-244.
[ Links ]
Williams, WT; Lambert, JM. 1966. Multivariate methods in plant ecology. V. Similarity analysis and informationanalysis. Journal of Ecology 54(2):427-445. [ Links ]
Wright, S. 1978. Evolution and the genetics of populations. Vol. 4 Variability within and among natural populations. University of Chicago Press, Chicago, USA. 580 p. [ Links ]
Yule, GU. 1911. An introduction of the theory of statistics. Charles Griffin & Company, Londres, Inglaterra. 376 p. [ Links ]
*Correspondencia a: Carlos Alberto Núñez-Colín & Diana Escobedo-López: Campo Experimental Bajío, Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias. Km. 6.5 Carretera Celaya – San Miguel de Allende. Apartado Postal 112. Celaya, 38010, Guanajuato, México. lit007a@gmail.com; escobedo.diana@inifap.gob.mx.
1 Proyecto Actualización en metodologías estadísticas para la gestión de recursos fitogenéticos del INIFAP, México.
2 Campo Experimental Bajío, Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias. Km. 6.5 Carretera Celaya – San Miguel de Allende. Apartado Postal 112. Celaya, 38010, Guanajuato, México. lit007a@gmail.com; escobedo.diana@inifap.gob.mx.
Recibido: 30 de mayo, 2011. Aceptado: 3 de octubre, 2011.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}