










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Cardiología
Print version ISSN 1409-4142
Rev. costarric. cardiol vol.11 n.2 San José Dec. 2009
Choque cardiogénico: Historia, fisiopatología e implicaciones terapeúticas. Parte I
Fernando Zeledón Sa*, Edgar Méndez Ja, Juan Pucci Ca, Carlos Escalante Gb; Carlos Estrada Gc
a. *Servicio de Cirugía de Tórax y Cardiovascular, Hospital México, 7o piso, La Uruca, San José, Costa Rica. Tel. (506) 2242-6646; fzszeledon@gmail.com
b. Servicio de Ginecología
c. Hospital San Juan de Dios, San José, Costa Rica.
Resumen
El choque cardiogénico es la principal causa de muerte entre los pacientes que presentan un sindrome coronario agudo. Se presenta una revision histórica y conceptos fisiológicos y fisiopatológicos de esta condición clínica que son la base para su abordaje terapéutico, principalmente la revasculrización de emergencia con angioplastia o cirugía.
Palabras clave: choque cardiogénico, infarto agudo miocardio, angioplastía, cirugía de bypass coronario.
Abstract
Cardiogenic shock: History, pathophysiology and therapeutic implications. Part I.
Cardiogenic shock is the major cause of death among patients with acute coronary syndrome. The history as well as physiologic and pathophysiologic analysis of this clinical condition, based on its therapeutic management, are presented. This management predominantly involves urgent coronary angioplasty/stent placement or coronary artery bypass surgery.
Keywords: cardiogenic shock, acute myocardial infarction, angioplasty, coronary artery bypass graft.
Introducción
El choque cardiogénico (CC) es la principal causa de muerte en los pacientes que sufren un infarto agudo de miocardio (IAM)1. A pesar de los avances científicos y nuevas guías en el manejo de esta patología, su mortalidad sigue siendo muy alta. Desde inicio de los años 70 del siglo XX, comenzó la cirugía de bypass coronario de urgencia en pacientes con CC y 10 años después, la revascularización percutánea coronaria (RPC); ambos son los métodos terapéuticos de elección en este caso, según la Asociación Americana de Cardiología1. No obstante, pareciera que esto sólo está escrito en el papel, porque en la práctica se presenta otro escenario, y es por lo tanto necesario crear conciencia entre los médicos que manejan pacientes con CC.
Cualquier acto terapéutico tiene su sustento en el conocimiento de la fisiopatología de la enfermedad, la cual conlleva al desarrollo de estudios clínicos que apoyarán o desaprobarán el tratamiento en cuestión. En este artículo se revisará la historia y la fisiopatología del CC, lo cual ha dado por resultado un giro en el manejo de esta condición y ha disminuido la mortalidad desde un 80-90% hasta alrededor del 50% en la actualidad, en países industrializados. Conforme se difundan estas técnicas agresivas de tratamiento, que atacan el origen de la disfunción orgánica en forma rápida y el personal médico se instruya en la prevención del choque, es probable que la mortalidad de esta patología continúe disminuyendo.
Definición
La definición de CC se basa en un conjunto de parámetros hemodinámicos y datos clínicos que se originan de la hipoperfusión tisular1. Entre los primeros, se le presta mayor atención a los valores de presión arterial, ya sea la sistólica (PAS), cuando se encuentra por debajo de 80 mmHg o bien, una disminución en 30 mmHg en la presión arterial media (PAM) con respecto a la presión media basal, durante al menos 30 min de evolución2,3. Es importante, sin embargo, correlacionar lo anterior con un adecuado estado volumétrico, siendo la ecocardiografía doppler, un instrumento últil para valorar las presiones de llenado ventricular1. Otros aspectos hemodinámicos que se consideran en la definición de CC son: presión telediastólica ventricular izquierda mayor de 18 mmHg o derecha mayor de 15 mmHg e índice cardiaco menor de 1,8 L/min/m2; estos últimos parámetros necesitan métodos invasivos, como por ejemplo, la cateterización de la arteria pulmonar; y por lo tanto, no siempre se dispone de ellos para realizar el diagnóstico de CC. Entonces, la mayoría de veces, el diagnóstico se realiza en base al cuadro clínico del paciente; las manifestaciones de hipoperfusión sistémica más frecuentes son la alteración del estado de conciencia, oliguria, piel fría y húmeda1,2,3,4. Las cifras de presión arterial no constituyen un diagnóstico categórico de CC y es necesario valorar el contexto farmacológico y clínico que rodea al paciente. Por ejemplo, un paciente normotenso y con datos clínicos de hipoperfusión, pero bajo soporte vasoconstrictor e inotrópico, puede calificar como tal; por otro lado, un individuo con historia de insuficiencia cardiaca severa e hipotenso (por debajo de las cifras tensionales citadas), pero sin manifestaciones clínicas de isquemia tisular, no se encuentra en CC5,6.
Choque cardiogénico. Una perspectiva histórica
La palabra "choque" se usó por primera vez en 1743, para describir una condición moribunda después de un trauma severo7. Algunos años después, en 1768, William Heberden se refirió a la angina como un "desorden de la respiración"8. En 1880, Carl Weight notó que muchos pacientes que sufrían un IAM, presentaban trombosis coronaria y aterosclerosis; seguidamente, en 1881, Samuelson fue el primero en describir las manifestaciones clínicas del IAM que conllevaba a un colapso circulatorio8. En 1903, un profesor de fisiología holandés llamado William Einthoven, diseñó el electrocardiógrafo, lo cual permitió analizar la actividad eléctrica cardiaca. En aquél tiempo, casi todos los casos de IAM se diagnosticaban postmorten; sin embargo, en 1910 Obrastzow y Straschesko presentaron dos casos de IAM que fueron diagnosticados antes de que murieran8 y dos años después, James Eric presentó un reporte en el cual se ligaba la clínica de pacientes con IAM con las autopsias; él propuso que el IAM no siempre era inmediatamente fatal por lo que tenían que realizarse todos los esfuerzos posibles para diagnosticar y tratar a estos pacientes antes de que desarrollen el colapso cardiovascular y la muerte. La descripción de un electrocardiograma en un paciente con un IAM fue hecha por Pardee en 1920, lo cual fue de vital importancia en el futuro diagnóstico de esta condición patológica y sus consecuencias.
En 1935 y en 1940, Harrison y Blalock fueron los primeros en clasificar el choque de acuerdo a sus causas, tales como cardiogénico, hipovolémico, vasogénico y neurogénico. En 1934, Fishberg describió el comportamiento hemodinámico del choque cardiogénico, estableciendo las diferencias entre un paciente con CC debido a un IAM, de un paciente con una estenosis mitral e insuficiencia cardiaca congestiva8. Al inicio de los años 50 se publicaron series de trabajos de pacientes con CC, en los cuales se concluyó que la mortalidad oscilaba entre un 80% a 90%. En 1952, Agress sugirió una definición de CC a partir de sus trabajos en perros: "una reducción de aproximadamente un 30% en la presión arterial media, con mantenimiento de esta reducción por al menos 30 minutos, con evidencia electrocardiográfica de isquemia y ausencia de arritmias que explicaran la hipotensión"8; Binder, en 1955 propuso una definición clínica en humanos que establecía que la presión arterial sistólica debía de disminuir a menos de 80 mmHg por más de 30 minutos, la frecuencia cardiaca aumentar a más de 110 latidos/minutos y el paciente presentar signos de colapso circulatorio, tales como alteración del sensorio, cianosis, piel sudorosa y fría y oliguria. El tratamiento inicial del CC a inicios de los años 50, consistió en administrar oxígeno suplementario, flebotomías, morfina, vapor de alcohol etílico, digitálicos, quinidina, cortisol y sangre o plasma, en la mayoría de casos. En 1954, Griffith y cols, evaluaron el uso de vasoconstrictores, inotrópicos y cronotrópicos positivos tales como el isoproterenol, la metoxamina y la norepinefrina. La única de estas que mejoraba la condición clínica en pacientes con CC fue la norepinefrina, y por lo tanto, fue denominada como el fármaco de elección en el CC. No obstante, la mortalidad oscilaba alrededor del 100%8. Al inicio de los años 60, se introdujeron las unidades de cuidado coronario como áreas especializadas en el manejo de los pacientes con CC y en 1962, Mason Sones introdujo la angiografía coronaria selectiva en la Cleveland Clinic, en Ohio, lo cual permitió evaluar la anatomía coronaria de los pacientes con IAM8.
En 1962, Moulopoulos y cols diseñaron un tubo de polietileno de 20 cm que se insertaba en la aorta descendente en perros y se inflaba con CO2 durante la diástole, sincronizado con el electrocardiograma. Seguidamente, en 1968, Kantrowitz y col utilizaron un balón intraaórtico que se inflaba con helio en diástole en 5 pacientes con CC, todos los cuales mejoraron sus parámetros hemodinámicos. En 1971, Page y col postularon que se tenía que infartar alrededor de un 30 a 40% del miocardio para que se desarrolle CC, aunque en otros estudios no resultó importante el tamaño de área necrosada8. Mueller y cols enfatizaron que el principal objetivo del tratamiento del CC era mejorar la oxigenación del miocardio y establecieron que, tanto la norepinefrina como la dopamina, mejoran la perfusión periférica a expensas de un incremento en el consumo de oxígeno miocárdico, lo cual es nocivo para el miocardio isquémico y recomendaron ser cautos con el uso de estos fármacos. Dunkman y cols realizaron cirugías de bypass coronario de emergencia con el uso de circulación extracorpórea (desarrollada por Gibbon en 1953) en 15 pacientes con IAM y CC que no habían mejorado con el balón de contrapulsación intraórtica (BCIA); de ellos, 6 sobrevivieron, demostrando por primera vez que la revascularización quirúrgica de emergencia es útil en estos pacientes. Leinbach, en 1973, también sugirió que la revascularización quirúrgica de emergencia, en el contexto de pacientes con choque, era factible.
En 1977, Johnson y cols recomendaron la revascularización temprana (< 12 horas del inicio del IAM) aunada al uso del BCIA en el período postoperatorio. Mundth y cols también sugirieron la cirugía de bypass coronario de emergencia en pacientes tratados con BCIA que no mejoraban su índice cardiaco por encima de 2 L/min/m2. DeWood en 1980 comparó el tratamiento con BCIA asociado a cirugía de bypass coronario versus solamente BCIA, y aunque la mortalidad hospitalaria fue similar, la mortalidad a largo plazo fue menor en el grupo quirúrgico; además, demostró que la mortalidad fue menor en los pacientes que se revascularizaron dentro de las 16 horas del inicio de los síntomas de IAM. En los años 80’s también se estableció el uso de trombolíticos en el IAM con elevación del segmento ST, después de la publicación de los resultados de los estudios GISSI e ISIS-28. En 1998, Bowers y cols reportaron resultados favorables con la intervención coronaria percutánea de emergencia en pacientes con oclusión de la arteria coronaria derecha e infarto inferior y del ventrículo derecho8. En 1999, se publicó el estudio denominado SHOCK1, que iría a cambiar la visión del CC por parte de muchos médicos alrededor del mundo; fue el primer ensayo prospectivo y aleatorizado en pacientes con CC, que estableció la revascularización temprana, ya sea quirúrgica o percutánea como terapia de elección, como se verá más adelante.
Etiología del Choque Cardiogénico
La principal causa de CC es el IAM2; su incidencia oscila entre un 7 a 9%, predominando en el IAM con elevación del segmento ST (5 a 8%) en el cual se presenta tempranamente (hora 7-9), en comparación al IAM sin elevación de dicho segmento (2,5%, hora 76)1,2,9, aunque la mortalidad es similar en ambos2,10,11. Aproximadamente el 70% de los pacientes desarrollan el CC una vez que han sido hospitalizados, en comparación al 30% que lo presenta en la sala de urgencias2,12. Aunque usualmente el CC se asocia con el IAM, en realidad su etiología puede ser cualquier otro defecto cardiaco que afecte la función contráctil miocárdica en una situación extrema, como los estados finales de las cardiomiopatías, valvulopatías, miocarditis, contusión miocárdica, cirugía bajo circulación extracorpórea prolongada, mixoma en la aurícula izquierda, entre otros2,6. Por otro lado, aunque el CC asociado al IAM se acompaña de una función sistólica ventricular izquierda por lo general deprimida, también existe el síndrome del choque cardiogénico con función sistólica conservada, en el cual, la presentación clínica es semejante al choque con disfunción sistólica13. Desde el punto de vista fisiológico, la fracción de eyección del ventrículo izquierdo corresponde al volumen sistólico dividida entre el volumen telediastólico (VS/VTD)14; por lo tanto, si ambos factores disminuyen o aumentan, la fracción de eyección tenderá a permanecer relativamente normal, aunque en realidad el gasto cardiaco se encuentre disminuido. Existen causas agudas y crónicas de CC con función sistólica conservada. Entre las primeras, cabe mencionar el taponamiento cardiaco masivo, la insuficiencia aguda severa de las válvulas mitral y aórtica, el tromboembolismo pulmonar masivo y el infarto del ventrículo derecho. Entre las causas crónicas destacan la insuficiencia cardiaca diastólica en fases avanzadas, la estenosis crítica de la válvula mitral, la pericarditis constrictiva y las miocardiopatías restrictiva e hipertrófica13. Por último, el CC puede desencadenarse después de un IAM por causas iatrogénicas, entre las que destacan el uso inadecuado de los betabloqueadores (inhiben la taquicardia compensadora), inhibidores de la enzima convertidora de angiotensina (vasodilatación extrema), diuréticos (disminuyen el volumen circulante efectivo y por lo tanto, precarga y gasto cardiaco), morfina (disminución en exceso de la precarga) y el sobreuso de la hidroterapia en el infarto ventricular derecho (distensión de cámaras derechas con desplazamiento del septum interventricular hacia la izquierda y disminución del llenado ventricular izquierdo). Lo anterior no significa que estos medicamentos (o maniobras) no deban usarse, sino que su administración debe ser cautelosa y guiada por un análisis fisiopatológico de la condición del paciente, aspecto muy importante en la prevención del CC1,15.
Fisiopatología del choque cardiogénico: Una falla multiorgánica.
Hemodinamia y metabolismo miocárdico durante el CC
Durante un IAM, una placa ateromatosa sufre una fisura, disrupción, erosión y exposición de la matriz vascular, provocando activación plaquetaria e incremento de la expresión de moléculas de adhesión: las más involucradas son las integrinas plaquetarias GPIIb/IIIa, GPIb/IX y la GPIa/IIa, que se unen a sus receptores en el fibrinógeno, colágeno y en el factor Von Willebrand sobre la matriz extravascular. También se expresan otros receptores como el Sialil Lewis x, CD11b/CD18 y el CD11a/CD18 en los neutrófilos polimorfonucleares que se unen con la molécula de adhesión intercelular (Intercellular Cell Adhesion Molecule, en inglés) del endotelio vascular y de la superficie de las plaquetas. El trombo intravascular que finalmente resulta, se encuentra, por lo tanto, adherido al lumen de la arteria coronaria, obstruyendo el flujo sanguíneo, con pérdida de energía cinética sanguínea y turbulencia postestenótica16,17.
Jean Louis Marie Poiseuille (1838), médico fisiólogo francés y Gotthilf Heinrich Ludwing Hagen (1839), físico alemán, propusieron la ley de Poiseuilli-Hagen, que establece que:
Q = ΔPπr4/8lη
donde Q es el flujo, ΔP es el gradiente de presiσn, r es el radio, l es la longitud del tubo y η es la viscosidad del líquido18. La ley formulada por George Simon Ohm en 1827, para los circuitos eléctricos establece:
R= V / I
donde R es resistencia, V es la diferencia de potencial eléctrico (en Voltios) e I es la intensidad de la corriente (en amperios). La misma ecuación en términos cardiovasculares es:
R = ΔP / GC
donde ΔP es el gradiente o diferencia de presiones y GC es el flujo o gasto cardiaco. Unificando la ley de Poiseuille y la de Ohm, aplicadas a la resistencia del circuito cardiovascular, se obtiene:
R = 8lη / πr4
La ley de Poiseuille sólo es válida para tubos rígidos, cilíndricos, con un radio constante, líquidos incompresibles, con flujo laminar, viscosidad constante y un flujo no pulsátil; aunque estas situaciones no se cumplen en el sistema circulatorio, lo cierto es que desde el punto de vista fisiológico y fisiopatológico, ha contribuido enormemente a comprender la hemodinamia humana. Por ejemplo, ante una estenosis coronaria, disminuye el flujo distal a la obstrucción y aumenta la resistencia a la cuarta potencia; es decir, que mínimos cambios en el descenso de la luz (radio) coronaria provocan grandes cambios a nivel de la perfusión miocárdica, aún cuando predomine la vasodilatación por incremento en la demanda metabólica de los miocitos isquémicos17,19,20. Además, a diferencia de otros órganos, el corazón presenta una extracción de oxígeno del 75% en reposo, contra el 25% en otros tejidos, lo que implica que mayores demandas metabólicas de oxígeno sólo se pueden lograr aumentando el flujo sanguíneo coronario; los demás tejidos incrementan la extracción de oxígeno arterial20. Si durante el IAM no se cuenta con un adecuado abastecimiento de sangre por las arterias coronarias colaterales, se establece la necrosis isquémica y, dependiendo de la extensión de miocardio comprometido o de la viabilidad del preexistente, la posibilidad de aparición de un CC es inminente. La mayoría de vasos colaterales no son realmente funcionales en el ser humano y presentan un diámetro menor a 200 micrómetros16. La disminución del flujo sanguíneo coronario desencadena eventos predecibles: el gasto cardiaco disminuye, aumenta la presión telediastólica ventricular y la presión venosa pulmonar, produciéndose congestión en este órgano, lo que disminuye la saturación arterial de oxígeno (SAO2) de la hemoglobina y disminuye la presión de oxígeno arterial disuelto (PaO2) a nivel periférico. La alteración resultante en el gradiente de presión de O2 sangre arterial(100 mmHg)/mitocondria(< 1 mmHg), limita el metabolismo aeróbico del miocardio16. Existe además una relación no lineal entre la saturación venosa del oxígeno (SVO2) y el índice cardiaco (Fig. 1).
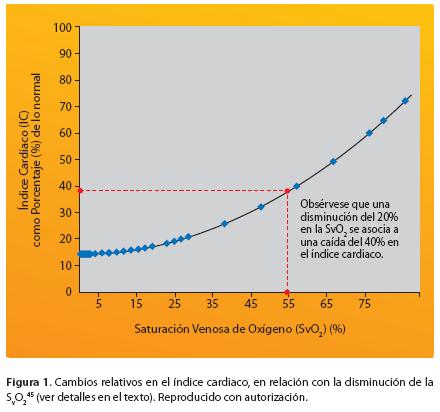
En condiciones fisiológicas, el metabolismo miocárdico depende principalmente (60-90%) de la oxidación de ácidos grasos (AG), ya que estos aportan mayor cantidad de adenosín trifosfato (ATP) por cada mol de AG metabolizado con respecto la oxidación de la glucosa. Por ejemplo, el uso del palmitato produce 105 moléculas de ATP en comparación a 32 ATP producidas cuando se utiliza la glucosa en condiciones aeróbicas21,22. Además, cuando se utiliza lípidos, se excluye el uso de carbohidratos y viceversa, con el objetivo de equilibrar la tasa metabólica con el consumo de oxígeno miocárdico (mVO2) y no utilizar dos vías metabólicas que requieren oxígeno al mismo tiempo21. El ATP se utiliza principalmente en la contracción muscular (60-70%), bombas iónicas (10-25% en especial, la Na+-K+ ATPasa y bombas de calcio) y en funciones diversas (5-30%)23,24. Durante el CC, se produce una hiperestimulación del sistema nervioso simpático, aumento de las catecolaminas circulantes, y de las hormonas de "estrés" (glucagón, cortisol, hormona de crecimiento), que estimulan la lipasa sensible a hormonas en el tejido adiposo y la lipoprotein lipasa en las células endoteliales y miocárdicas, las cuales incrementan la concentración de AG libres en el torrente sanguíneo; también se produce expresión de las proteínas que median su difusión facilitada en el sarcolema del cardiomiocito25. Debido a la alta tasa catabólica de ATP durante la isquemia, se produce un incremento en la concentración de adenosín monofosfato cíclico (AMP) y de proteincinasa estimulada por el AMP, la cual fosforila e inhibe a otra proteína enzimática, la acetil CoA carboxilasa, la que en condiciones fisiológicas se encarga de formar malonyl-CoA a partir de acetil-CoA21,25,26. El malonyl-CoA es el principal inhibidor de la oxidación de AG a nivel mitocondrial, ya que produce un importante descenso en la expresión de la palmitoil-cartinina-transferasa I, paso limitante de dicho proceso. Por lo tanto, al disminuir las concentraciones citoplasmáticas de malonyl-CoA se produce indirectamente un incremento en la oxidación de AG, al menos durante la isquemia miocárdica leve a moderada o en las etapas iniciales del CC21,25,26. Aunque los AG libres son el sustrato metabólico preferido por el miocardio en condiciones de aerobiosis, durante la isquemia su uso se convierte en un factor nocivo para la homeostasis celular cardiaca, ya que su oxidación requiere de una mayor tasa de mVO2 en comparación al uso de carbohidratos25,26; por lo tanto, esto pone en desventaja a un tejido miocárdico que lo que más bien requiere, es disminuir el mVO2 ante aportes de oxígeno limitados. Además, parte de los AG no se pueden metabolizar y se acumulan en el citosol en forma libre o de triglicéridos, lo que promueve la apertura de canales de calcio en el sarcolema, fomentando una sobrecarga de calcio, una mayor degradación de ATP por parte de las bombas de calcio, inestabilidad de la membrana plasmática y generación de arritmias23,25,26,27.
En esta fase se encuentra bloqueada la vía glucolítica aeróbica, ya que la acumulación de acetil-CoA, dinucleótido de favina-adenina (FAD) reducido y el dinucleótido de nicotinamida adenina (NAD) reducido como productos de varias reacciones metabolíticas (entre ellas, el paso de lactato a piruvato por la enzima lactato deshidrogenasa), es un potente inhibidor de la piruvato deshidrogenasa, enzima que se encuentra en la membrana mitocondrial interna y que se encarga de la descarboxilación, acetilación y deshidrogenación del piruvato para convertirlo en acetil-CoA, el cual posteriormente ingresa a la vía del ciclo de Krebs y la glucólisis aeróbica. Conforme continúa disminuyendo el aporte de oxígeno a la mitocondria, los procesos aeróbicos se ven limitados, incluyendo la oxidación de AG (que requiere de FAD y NAD en su forma oxidada), disminuye la concentración de citrato (intermediario de ciclo de Krebs) y se incrementan aún más el AMP y los fosfatos inorgánicos. Todos ellos estimulan la acción de la fosfofrutokinasa-1 (enzima estimulante de la glucólisis anaeróbica) y la fosfofrutokinasa-2, que ejerce una retroalimentación positiva sobre la primera21,25,26. Por lo tanto, durante la evolución de la isquemia se evidencian dos fases: en una etapa temprana se promueve la utilización de AG y en una segunda fase, la utilización anaeróbica de carbohidratos, debida a las circunstancias del microambiente28. Esto representa algunas ventajas metabólicas para el cardiomiocito isquémico, en especial en lo que respecta a la disminución de la demanda de oxígeno, ya que los AG generan un mVO2 18% mayor que cuando se utiliza la glucosa en condiciones aeróbicas, brecha que incrementa aún más en situaciones de anaerobiosis21,23,25.
El aumento de la difusión facilitada de glucosa durante la isquemia se debe a un incremento en la expresión y translocación de los transportadores de glucosa GLUT-1 y GLUT-4 a nivel sarcolémico, promovidos por la fosforilación efectuada por la proteincinasa estimulada por el AMP25,29,30. La piruvato deshidrogenasa continúa inhibida debido al acúmulo de NAD reducido, al incremento de CO2 tisular y a la deficiencia de oxígeno. El piruvato entonces se metaboliza a lactato por acción de la lactato deshidrogenasa, el cual puede ser exportado por un transportador monocarboxílico situado en el sarcolema miocárdico. Esta reacción es bidireccional, pero en condiciones de isquemia severa, se encuentra desplazada hacia la formación de lactato21. Como se ha mencionado, la glucólisis es de predominio anaeróbica, ya que el piruvato no se puede degradar a acetil CoA e ingresar en el ciclo de los ácidos tricarboxílicos o de Krebs, lo que disminuye abismalmente la producción energética a 2 moléculas de ATP por mol de glucosa, apenas un 6% del obtenido del metabolismo oxidativo23. La regeneración de compuestos de alto valor energético a partir de la fosfocreatina también se ve deteriorada; el glucógeno fue rápidamente metabolizado y el líquido intracelular comienza a tornarse acidótico23. Cabe señalar que una "acidosis intracelular leve" no es nociva para el medio interno celular miocárdico, ya que disminuye el trabajo miocárdico en aras de equilibrar la relación entre el aporte/demanda miocárdica de oxígeno28. Aunque el aporte energético de la glucólisis anerobia pareciera ser suficiente para mantener la homeostasia de la membrana plasmática, la progresión de la acidosis celular termina por inhibir la glicólisis y la producción de ATP21,23. Además, los hidrogeniones compiten con el calcio para unirse a la troponina C, desacoplando el proceso de excitación-contracción28.
Aunque la mayoría de textos señalan a la producción de lactato como la causa de la acidosis intracelular, algunos autores21,28 sostienen que se debe más bien al consumo exagerado de ATP, tal como se evidencia por la siguiente reacción:
Glucosa + 2 Mg-ADP + 2 Pi2-- ------- 2 lactato- + 2 Mg-ATP2-
(en condiciones aeróbicas)
2 Mg-ATP2- ------- 2 Mg-ADP- + 2 Pi 2- + 2 H+
(en condiciones anaeróbicas)
donde Mg-ADP es adenosin difosfato magnésico, Pi2- es fosfato inorgánico, Mg-ATP2- es adenosin trifosfato magnésico oxidado y H+ es ión hidrogenión. Debido a la desproporción de cargas en la reacción, se generan 2 iones H+, contribuyendo así a la génesis y perpetuación de la acidosis, a menos que se instaure una terapeútica que restablezca el aporte de oxígeno.
En condiciones fisiológicas, la osmolaridad entre los compartimentos extra e intracelular se encuentra en equilibrio, de manera tal que no existe un gradiente osmótico importante que comprometa la integridad de las funciones celulares. Sin embargo, durante la isquemia prolongada, la cinética de la bomba Na+-K+ ATPasa disminuye y se incrementa la concentración de sodio intracelular, el contratransportador Na+-Ca++ (que en condiciones fisiológicas expulsa calcio) limita su función, o inclusive transporta calcio en sentido inverso al líquido intracelular, promoviendo, ambas situaciones, un aumento de la concentración de calcio intracelular28. Además, el contratransportador Na+-H+ incrementa su actividad (es decir, expulsa hidrogeniones en intercambio con sodio), lo que fomenta aún más el aumento de la concentración de sodio en el líquido intracelular del cardiomiocito28. Otro mecanismo que amortigua la elevada concentración de hidrogeniones en el líquido intracelular es la lanzadera malato-aspartato, la cual, en condiciones fisiológicas, transporta el NAD reducido y los hidrogeniones hacia la mitocondria, para ser utilizados en la cadena respiratoria, ya que son impermeables a la membrana mitocondrial21,28. Esta lanzadera disminuye su actividad durante la isquemia, lo que acelera la acidosis celular. Durante la reperfusión, la pérdida de funcionalidad de esta lanzadera se ha asociado a la génesis del aturdimiento miocárdico28; la concentración de H+ en el líquido intracelular también es incrementada por la inhibición de la piruvato deshidrogenasa, como se ya se ha mencionado. La sobrecarga de sodio, calcio, hidrogeniones, potasio, alanina, lactato, succinato, adenosina y otros desechos metabólicos producen incremento de la osmolaridad del líquido intracelular, ósmosis subsecuente y edema celular, contribuyendo a la muerte de los miocitos por apoptosis promovida por la liberación del citocromo C mitocondrial, oncosis (necrosis) o autofagia; esta última es desencadenada por la activación de las proteasas dependientes de calcio y la lisis de los lisosomas, con la liberación de más enzimas autofágicas16.
La disfunción sistólica y diastólica del miocardio conlleva a una reducción de su distensibilidad y por ende a su dilatación; además, según la ley de Laplace (1805):
Tensión = presión x radio / grosor
aumenta la tensión parietal, la cual incrementa en proporción directa el mVO232. Los eventos concomitantes tales como taquicardia (que aumenta el mVO2 y disminuye el tiempo de diástole), bradicardia excesiva (que incrementa la distensión y disminuye el gasto cardiaco) y la hiperactividad del sistema nervioso simpático, sistema renina-angiotensina-aldosterona y vasopresina (que aumentan la poscarga) contribuyen adicionalmente al deterioro de la función miocárdica23 (Fig. 2).
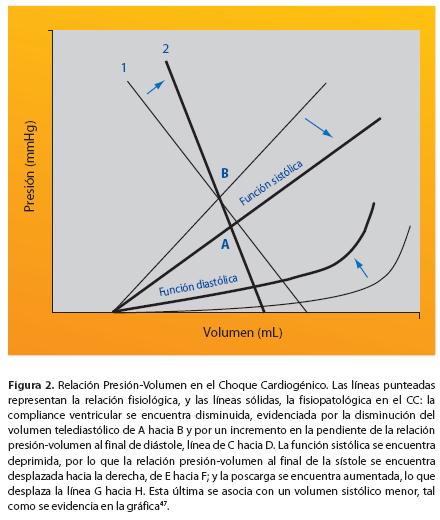
La presión telediastólica está aumentada, a menos que exista hipovolemia concomitante, y el miocardio se encuentra trabajando en la porción descendente de la curva de Frank-Starling23. Tal elevación en el ventrículo izquierdo se traduce en un aumento de la presión atrial izquierda y en un aumento de la presión hidrostática intersticial pulmonar, lo que conlleva a edema pulmonar, alteración en la relación ventilación/perfusión e hipoxemia6. La presión venosa central por lo general se encuentra aumentada y la presión aórtica disminuida, por lo que el gradiente para el flujo coronario disminuye, empeorando aún más la perfusión miocárdica. La resultante disminución del gasto cardiaco conlleva a una hipoperfusión generalizada y disfunción multiorgánica.
El cerebro durante el CC
Cuando la PAM desciende por debajo de 50 mmHg, se pierde la autorregulación del flujo sanguíneo cerebral y este se hace dependiente de la presión de perfusión, la cual en el CC está disminuida, desencadenando en isquemia cerebral generalizada, alteraciones en la membrana neuronal y manifestaciones clínicas como somnolencia o alteración del estado de conciencia23.
El riñón durante el CC
Durante las etapas iniciales del CC y cuando la PAM se encuentra por debajo de 80 mmHg, la activación de los barorreceptores localizados en la arteriola aferente y la menor entrega de cloruro de sodio a la mácula densa, estimulan a las células granulosas para que secreten renina, la cual, en última instancia, incrementa la concentración de angiotensina II y aldosterona, promoviendo la reabsorción de sodio y agua y la secreción de potasio e hidrogeniones a nivel tubular33. El consiguiente aumento de la volemia produce un incremento del retorno venoso cardiaco, en aras de mejorar el gasto cardiaco, según la ley de Frank-Starling. No obstante, lejos de alcanzar dicho objetivo, en el IAM del ventrículo izquierdo, este incremento de la precarga fomenta distensión de la fibra miocárdica, dilatación, aumento de las presiones al final de diástole, lo que conlleva a un desequilibrio entre el aporte y demanda de oxígeno. La insuficiencia renal aguda es una de las complicaciones graves del CC, instaurándose dentro de las primeras 36-72 horas de iniciado23; se produce disminución del flujo sanguíneo cortical, isquemia de las células tubulares y después necrosis, obstrucción tubular y edema intersticial23.
El pulmón durante el CC
El edema pulmonar y la disminución de la distensibilidad de este órgano produce un aumento en el espacio muerto fisiológico, alteración de la relación V/Q y elevación del gradiente de oxígeno alveólo-capilar. La hipoxia alveolar provoca vasoconstricción de las arteriolas pulmonares e incremento de la resistencia vascular, lo que se traduce en un aumento de la poscarga al ventrículo derecho y disminución del contenido arterial de oxígeno. La hipoxemia y el incremento de la concentración de hidrogeniones plasmáticos estimulan a los quimiorreceptores periféricos, lo que induce hiperventilación (respiración de Kussmaul) y agotamiento de la musculatura ventilatoria.
El tracto gastrointestinal durante el CC
La hiperestimulación simpática provoca una redistribución del flujo sanguíneo desde los órganos que poseen una rica expresión de receptores alfa, hacia el corazón y sistema nervioso central. El tracto gastrointestinal sufre isquemia, aumento de la permeabilidad celular y translocación bacteriana23. Debido al gradiente de presión reducido entre el sistema porta y la vena hepática (normal 5-8 mmHg), al aumentar la presión en el atrio derecho el hígado se congestiona y ocurre edema celular y necrosis de células hepáticas4,5; disminuye su capacidad de interconvertir lactato en piruvato y glucosa (fenómeno conocido como ciclo de Cori) y de sintetizar glucógeno (glucogénesis), contribuyendo de esta forma a la hiperlactatemia4,5.
El sistema hemato-inmunológico durante el CC
Durante el infarto se produce inflamación aguda miocárdica, con vasodilatación y exudado rico en proteínas del tejido dañado34. Esta vasodilatación es propiciada por la síntesis y secreción de óxido nítrico por las células endoteliales y los leucocitos, por la liberación de mediadores lipídicos (prostaglandinas) a partir del ácido araquidónico, a través de la acción de la ciclooxigenasa y por otros estímulos35. Los neutrófilos activados presentan marginación, adhesión y migración hacia el tejido inflamado por la interacción con las moléculas de adhesión, tales como selectinas e integrinas. Luego, estos leucocitos secretan quimioquinas (por ejemplo, la interleuquina-8 (IL), las cuales atraen aún más células inflamatorias al tejido en mención. La vía extrínseca de la coagulación se activa al expresarse el factor tisular en las células endoteliales y en monocitos activados en respuesta al factor de necrosis tumoral (FNT-alfa), IL-1, IL-6 y a la proteína C reactiva. La interacción del factor tisular con el complejo VIIa de la coagulación, induce la producción de citoquinas pro-inflamatorias, tales como el FNT-alfa por los macrófagos y células endoteliales. Por lo tanto, existe una estrecha relación entre la cascada de la coagulación y la respuesta inflamatoria35,36. También se produce una activación del complemento, exacerbando aún más la inflamación.
Las vías innata y adaptativa del sistema inmunológico también se hiperestimulan. La vía innata está dada por los macrófagos, células asesinas, neutrófilos, células endoteliales, complemento y cascada de la coagulación, además de sus citoquinas y factores proinflamatorios. La vía adquirida es mediada por los linfocitos T y B. A nivel de las céluas T, se promueve la reacción de tipo Th-1, que consiste en hiperestimulación de la inmunidad celular. Los macrófagos y células T secretan el FNT-alfa, el cual promueve la activación de neutrófilos, células endoteliales, fiebre (ejecutada por el hipotálamo), secreción de reactantes de fase aguda (por parte del hígado), catabolismo proteico y lipídico, depresión miocárdica, secresión de quimioquinas y apoptosis en varios tejidos. Además, se estimula el eje hipotálamo-hipófisis-glándulas suprarrenales. La IL-1 es principalmente secretada por los macrófagos y produce funciones semejantes al FNT-alfa. El interferón gamma es secretado por las células T y fomenta la activación de las células asesinas, macrófagos y células T y B. La IL-8 e IL-12 actúan de forma semejante, promoviendo la activación de macrófagos y células T, presentándose, por lo tanto, una retroalimentación positiva inmunológica35. En conclusión, durante el CC se promueve una respuesta inflamatoria e inmunológica a gran escala, que contribuye aún más en la instauración del círculo vicioso metabólico antes descrito, termina en falla multiorgánica y en la muerte del paciente.
El tejido muscular y piel durante el CC
La hipoperfusión del músculo y de la piel contribuyen a la hiperlactinemia. El transporte iónico del músculo esquelético se ve alterado, con desacople entre excitación-contracción. El diafragma es uno de los principales músculos afectados, disminuyendo su actividad y propiciando insuficiencia respiratoria23. La piel se torna fría y el llenado capilar disminuye, debido a la disminución de su irrigación sanguínea.
Presentaciónclínica
Una valoración rápida del paciente con CC es crítica para determinar la terapia subsecuente. La mayoría de pacientes (70%) desarrollan el choque durante su hospitalización y una minoría a su ingreso a la sala de emergencias. El paciente con CC puede presentarse con disnea debida a la disminución en la distensiblidad pulmonar y a la hipoxemia que resulta de la alteración de la relación ventilación/perfusión, ambas inducidas por el edema intersticial y alveolar. La hipoperfusión sistémica que produce la hipotensión debida a la disminución del gasto cardiaco conlleva alteraciones en el estado de conciencia, piel fría, húmeda, pulso rápido, débil (e irregular si se presentan arritmias) y oliguria. Puede además presentarse ingurgitación venosa yugular por insuficiencia ventricular izquierda o derecha y crepitaciones pulmonares -solo en la falla ventricular izquierda-. Los ruidos cardiacos se encuentran hipofonéticos, con un tercer o cuarto ruido cardiacos producidos por alteración de la relajación o de la distensibilidad ventricular respectivamente. Se puede auscultar un soplo holosistólico, debido a insuficiencia mitral o a ruptura del tabique interventricular), aunque algunas veces puede ser difícil auscultarlo, debido a la disminución del gradiente de presión entre las cámaras cardiacas y por ende, disminución en la turbulencia sanguínea2,6.
El CC debido a infarto del ventrículo derecho también puede presentarse con congestión pulmonar, debido al desplazamiento del tabique interventricular hacia el ventrículo izquierdo y el consiguiente aumento de las presiones de llenado izquierdas. Además, existe ingurgitación yugular paradójica con la inspiración, como consecuencia de la presión elevada en la aurícula derecha; en cambio, en condiciones fisiológicas, la presión intrapleural se hace más negativa, aumenta el retorno venoso a las cámaras derechas y las yugulares se "colapsan" durante la inspiración7,38.
Abreviaturas: AG: ácidos grasos; AMP: adenosín monofosfato cíclico; ATP: adenosín trifosfato; BCIA: balón de contrapulsación intraórtica; CC: choque cardiogénico; CoA: acetil coenzima A; DO2: Entrega de oxígeno; FAD: dinucleótido de favina-adenina; FNT: factor de necrosis tumoral; H+: ion hidrogenión; IAM: infarto agudo del miocardio; IL: interleukina; mVO2: consumo de oxígeno miocárdico; NAD: dinucleótido de nicotinamida-adenina; PAD: presión arterial diastólica; PAM: presión arterial media; PaO2: presión de oxígeno arterial disuelto; PAS: presión arterial sistólica: SAO2: Saturación arterial de oxígeno; SvO2: Saturación venosa de oxígeno; VS: volumen sistólico; VTD: volumen telediastólico.
Referencias
1. Reynolds H, Hochman J. Cardiogenic Shock: Current Concepts and Improving Outcomes. Circulation 2008; 117: 686-697. [ Links ]
2.Topalian S, Ginsberg F, Parrillo J. Cardiogenic Shock. Crit Care Med 2008; 36: S66-S74. [ Links ]
3. Ashby D, Stone G, Moses J. Cardiogenic Shock in Acute Myocardial Infarction. Catheterization and Cardiovascular Interventions 2003; 59: 34-43. [ Links ]
4. Alpert JS, Becker RC. Cardiogenic Shock: Elements of Etiology, Diagnosis, and Therapy. Clin. Cardiol 1993; 16: 182-190. [ Links ]
5. Hasdai D. Cardiogenic Shock. Contemporary Cardiology: Cardiogenic Shock: Diagnosis and Treatment. Cap 1: 3-32 [ Links ]
6. Hollenberg S, Kavinsky C, Parrillo J. Cardiogenic Shock. Ann Intern Med 1999; 131: 47-59. [ Links ]
7. Ginsberg F, Parrillo J. Cardiogenic Shock: A Historical Perspective. Crit Care Clin 2009; 25: 103-114. [ Links ]
8. Manji R, Wood k, Kumar A. The History and Evolution of Circulatory Shock. Crit Care Clin 2009; 25: 1-29. [ Links ]
9. Martinez-Rumayor A, Januzzi J. Non- ST Segment Elevation Acute Coronary Syndromes: A Comprehensive Review. Southern Medical Journal 2006; 99: 1103-1110. [ Links ]
10. Valente S, Lazzeri C, Chiostri M, et al. Time of onset and outcome of cardiogenic shock in acute coronary syndromes. Journal of Cardiovascular Medicine 2008; 9: 1235-1240. [ Links ]
11. Dar O, Cowie M. Acute heart failure in the intensive care unit: Epidemiology. Crit Care Med 2008; 36: S3-S8. [ Links ]
12. Goldberg R, Spencer F, Gore J, et al. Thirty-Year Trends (1975 to 2005) in the Magnitude of, Management of, and Hospital Death Rates Associated With Cardiogenic Shock in Patients With Acute Myocardial Infarction: A Population-Based Perspective. Circulation 2009; 119: 1211-1219. [ Links ]
13. Chatterjee K, McGlothlin D, Michaels A. Analytic Reviews: Cardiogenic Shock with Preserved Systolic Function: A Reminder. J Intensive Care Med 2008; 23: 355-366. [ Links ]
14. Guyton AC, Hall JE. Tratado de Fisiología Médica. Philadelphia: McGraw-Hill, 1997:115-129. [ Links ]
15. Barry W, Sarembock I. Cardiogenic Shock : Therapy and Prevention. Clin Cardiol 1998; 21: 72-80. [ Links ]
16. Okuda M. A Multidisciplinary Overview of Cardiogenic Shock. SHOCK 2006; 25: 557-570. [ Links ]
17. Braunwald E. Heart Disease. Philadelphia: W.B. Saunders Co., 1996: 1265-1290. [ Links ]
18. Little RC, Little WC. Physiology of the Heart and Circulation. St Louis: Mosby-Year Book: 219-243. [ Links ]
19. Boron W, Boulpaep EL. Textbook of Medical Physiology. Philadelphia: W.B. Saunders Co., 2002: 426-446. [ Links ]
20. Mohrman DE, Heller LJ. Fisiología Cardiovascular. México D.F.:McGraw-Hill, 2007:47-70. [ Links ]
21. Sperelakis N. Heart Physiology and Pathophysiology. Philadelphia: Academic Press., 2001: 543-565. [ Links ]
22. van der Vusse G, van Bilsen M, Glatz J. Cardiac fatty acid uptake and transport in health and disease. Cardiovascular Research 2000; 45: 279-293. [ Links ]
23. Williams S, Wright D, Tan L. Management of cardiogenic shock complicating acute myocardial infarction: towards evidence based medical practice. Heart 2000; 83: 621-626. [ Links ]
24. Marín-García J, Goldenthal M. Fatty acid metabolism in cardiac failure: biochemical, genetic and cellular analysis. Cardiovacular Research 2002; 54: 516-527. [ Links ]
25. Stanley W, Recchia F, Lopaschuk G. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol Rev 2005; 85: 1093-1129. [ Links ]
26. Stanley W, Lopaschuck G, McCormack J. Regulation of energy substrate metabolism in the diabetic heart. Cardiovascular Research 1997; 34: 25-33. [ Links ]
27. Lopaschuck G, Folmes C, Stanley W. Cardiac Energy Metabolism in Obesity. Circ Res 2007; 101: 335-347. [ Links ]
28. Taegtmeyer H, King L, Jones B. Energy Substrate Metabolism, Myocardial Ischemia, and Targets for Pharmacotherapy. Am J Cardiol 1998; 82: 54K-60K. [ Links ]
29. Schwenk R, Luiken J, Bonen A, et al. Regulation of sarcolemmal glucose and fatty acid trasnporters in cardiac disease. Cardiovascular Research 2008; 79: 249-258. [ Links ]
30. Ingwall J, Weiss R. Is the Failing Heart Energy Starved? On Using Chemical Energy to Support Cardiac Function. Circ Res 2004; 95: 135-145. [ Links ]
31. Patterson B, Fields A, Shannon R. New insights into myocardial glucose metabolism: surviving under stress. Curr Opin Clin Nutr Metab Care 2009; 12: 424-430. [ Links ]
32. Sadoshima J, Izumo S. The Cellular And Molecular Response of Cardiac Myocytes to Mechanical Stress. Annu Rev Physiol 1997; 59: 551-571. [ Links ]
33. Ulate G. Fisiología renal. San José: Editorial Universidad de Costa Rica, 2006. [ Links ]
34. Debrunner M, Schuiki E, Minder E, et al. Proinflammatory cytokines in acute myocardial infarction with and without cardiogenic shock. Clin Res Cardiol 2008; 97: 298-305. [ Links ]
35. Sherwood E. Mechanisms of the inflammatory response. Best Practice & Research Clinical Anaesthesiology 2004; 18: 385-405. [ Links ]
36. Kohl B, Deutschman C. The inflammatory response to surgery and trauma. Curr Opin Crit Care 2006; 12: 325-332. [ Links ]
37. Pfisterer M. Right ventricular involment in myocardial infarction and cardiogenic shock. Lancet 2003; 362:392-94. [ Links ]
38. Vlahakes G. Right ventricular failure following cardiac surgery. Coronary Artery Disease 2005; 16: 27-30. [ Links ]
39. Gogo P. The Evaluation and Management of Cardiogenic Shock. Crit Pathways in Cardiol 2006; 5: 1-6. [ Links ]
40. Gurm H, Bates E. Cardiogenic Shock Complicating Myocardial Infarction. Crit Care Clin 2007; 23: 759-777. [ Links ]
41. Antonelli M, Levy M, Andrews P, et al. Hemodynamic monitoring in shock and implications for management. International Consensus Conference, Paris, France 27-28 April 2006. Intensive Care Med 2007; 33: 575-590. [ Links ]
42. Pinsky M. Hemodynamic Evaluation and Monitoring in the ICU. Chest 2007; 132: 2020-2029. [ Links ]
43. Pinsky M. Rationale for cardiovascular monitoring. Curr Opin Crit Care 2003; 9: 222-224. [ Links ]
44. Pinsky MR, Payen D. Functional Hemodynamic Monitoring. New York: Springer 2005: 233-50. [ Links ]
45. Tibby S, Murdoch I. Monitoring cardiac function in intensive care. Arch Dis Child 2003; 88: 46-52. [ Links ]
46. Hoffman G, Ghanayem N, Tweddell J. Noninvasive Assessment of Cardiac Output. Semin Thorac Cardiovasc Surg Pediatr Card Surg Ann 2005; 8: 12-21. [ Links ]
47. Liepert D, Rosenthal M; Thomas S. Shock: A Comparative Physiologic Approach to Mechanisms and Therapy. Seminars in Anesthesia, Perioperative Medicine and Pain 1999; 18: 1-14. [ Links ]
48. Hett D, Jonas M. Non-invasive cardiac output monitoring. Current Anaesthesia & Critical Care 2003; 14: 187-191. [ Links ]
49. Kuper M. Continous cardiac output monitoring. Current Anaesthesia & Critical Care 2004; 15: 367-377.
50. Maus T, Lee D. Arterial Pressure-Based Cardiac Output Assessment. Journal of Cardiothoracic and Vascular Anesthesia 2008; 22: 468-473. [ Links ]
51. Gelman S. Venous Function and Central Venous Pressure. Anesthesiology 2008; 108: 735-748. [ Links ]
52. Madger S. Central venous pressure : A useful but not so simple measurement. Crit Care Med 2006; 34: 2224-2227. [ Links ]
53. Hadian M, Pinsky M. Functional hemodynamic monitoring. Curr Opin Crit Care 2007; 13: 318-323. [ Links ]
54. Fincke R, Hochman J, Lowe A, et al. Cardiac Power Is the Strongest Hemodynamic Correlate of Mortality in Cardiogenic Shock: A Report from the SHOCK Trial Registry. J Am Coll Cardiol 2004; 44: 340-348. [ Links ]
55. Hasdai D, Topol E, Califf R, et al. Cardiogenic shock complicating acute coronary syndromes. Lancet 2000; 356: 749-756. [ Links ]
56. Santa-Cruz R, Cohen M, Ohman E. Aortic Counterpulsation: A Review of the Hemodynamic Effects and Indications for Use. Catheterization and Cardiovascular Interventions 2006; 67: 68-77. [ Links ]
57.Tsagalou E, Anastasiou-Nana M, Nanas J. Intra-Aortic Balloon Counterpulsation for the Treatment of Myocardial Infarction Complicated by Acute Severe Heart Failure. Congest Heart Fail 2009; 15: 35-40. [ Links ]
58. Trost J, Hillis D. Intra-Aortic Balloon Counterpulsation. Am J Cardiol 2006; 97: 1391-1398. [ Links ]
59. Papaioannou T, Mathioulakis D, Stametelopoulos K, et al. New Aspects on the Role of Blood Pressure and Arterial Stiffness in Mechanical Assistance by Intra-aortic Balloon Pump: In-vitro Data and Their Application in Clinical Practice. Artif Organs 2004; 28: 717-727. [ Links ]
60. Burkhoff D, Cohen H, Brunckhorst C, et al. A randomized multicenter clinical study to evaluate the safety and efficacy of the TandemHeart percutaneous ventricular assist device versus conventional therapy with intraaortic balloon pumping for treatment of cardiogenic shock. Am Heart J 2006; 152 (469): el-8. [ Links ]
61.Hillis L, Lange R. Optimal Management of Acute Coronary Syndromes. N Engl J Med 2009; 360: 2237-2240. [ Links ]
62. Keeley E, Hillis L. Primary PCI for Myocardial Infraction with ST-Segment Elevation. N Engl J Med 2007; 356: 47-54. [ Links ]
63. Silber S, Albertsson P, Avilés F, et al. Guidelines for Percuatneous Coronary Interventions. The Task Force for Percutanenous Coronary Interventions of the European Society of Cardiology. European Heart Journal 2005; 26: 804-847. [ Links ]
64. Ducas J, Grech E. Percutaneous coronary intervention: cardiogenic shock. BMJ 2003; 326: 1450-1452. [ Links ]
65. Williams S, Wright D, Tan L. Management of cardiogenic shock complicating acute myocardial infarction: towards evidence based medical practice. Heart 2000; 83: 621-626. [ Links ]
66. Faxon D. Early reperfusion strategies after acute ST-segment elevation myocardial infarction: the importance of timing. Nature Clinical Practice Cardiovacular Medicine 2005; 2: 22-28. [ Links ]
67. Hochman JS, Sleeper L, Webb J, et al. Should We Emergently Revascularize Occluded Coronaries for Cardiogenicc Shock: An International Randomizadel Trial of Emergency PTCA/CABG-trial design. Am Heart J 1999; 137:313-21. [ Links ]
68. White H, Assmann S, Sanborn T, et al. Comparison of Percutaneous Coronary Intervention and Coronary Artery Bypass Grafting After Acute Myocardial Infarction Complicated by Cardiogenic Shock : Results From the Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock ( SHOCK ) Trial. Circulation 2005; 112: 1992-2001 [ Links ]
69. Jeger R, Harkness S, Ramanathan K, et al. Emergency revascularization in patients with cardiogenic shock on admission: a report from the SHOCK trial and registry. European Heart Journal 2006; 27: 664-670. [ Links ]
70. Patel M, Dehmer G, Hirshfeld J, et al. ACCF/SCAI/STS/AATS/AHA/ASNC 2009 Appropriateness Criteria for Coronary Revascularization. J Am Coll Cardiol 2009; 53: 530-553. [ Links ]
71. Morrow, D. Antithrombotic Therapy to Support Primary PCI. N Engl J Med 2008; 358: 2280-2282. [ Links ]
72. Serruys P, Kutryk M, Ong A. Coronary-Artery Stents. N Engl J Med 2006; 354: 483-495. [ Links ]
73. Mann H, Nolan P. Update on the management of cardiogenic shock. Curr Opin Crit Care 2006; 12: 431-436. [ Links ]
74. Sanborn T, Feldman T. Management strategies for cardiogenic shock. Current Opinion in Cardiology 2004; 19: 608-612. [ Links ]
75. Comas G, Esrig B, Oz M. Surgery for Myocardial Salvage in Acute Myocardial Infarction and Acute Coronary Syndromes. Heart Failure Clin 2007; 3: 181-210. [ Links ]
76. Raanani E, Avramovich A, Hasdai D, et al. Operative Strategies for CArdiogenic Shock Complicating Acute Coronary Syndromes. Contemporary Cardiology: Cardiogenic Shock: Diagnosis and Treatment. Cap 7 : 77-91 [ Links ]
77. Sundell J, Knuuti J. Insulin and Myocardial blood flow. Cardiovascular Research 2003; 57: 312-319. [ Links ]
78. Bertrand L, Horman S, Beauloye C, et al. Insulin signalling the heart. Cardiovascular Research 2008; 79: 238-248. [ Links ]
79. Brownsey R, Boone A, Allard M. Actions of insulin on the mammalian heart: metabolism, pathology and biochemical mechanisms. Cardiovascular Research 1997; 34: 3-24 [ Links ]
80. Metha SR, Yusuf S, Díaz R, et al. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: the CREATE-ECLA randomized controlled trial. JAMA 2005; 293(4):437-46. [ Links ]
81. Rastan AJ, Eckenstein JI, Hentschel B, et al. Emergency coronary artery bypass graft surgery for acute coronary syndrome: beating heart vs conventional cardioplejic cardiac arrest strategies. Circulation 2006; 114 (suppl I): 477-485. [ Links ]
82. Leopold, J. Does Thrombolytic Therapy Facilitate of Foil Primary PCI? N Engl J Med 2008; 358; 2277-2279. [ Links ]
83. Kierman T, Gersh B. Thrombolysis in Acute Myocardial Infarction: Current Status. Med Clin N Am 2007; 91: 617-637. [ Links ]
84. De Luca G, Suryapranata H, Marino P. Reperfusion Strategies in Acute ST-Elevation Myocardial Infarction: An Overview of Current Status. Progress in Cardiovascular Diseases 2008; 50: 352-382. [ Links ]
85. Ortolani P, Marzocchi A, Marrozzini C, et al. Clinical impacto from direct referral to primary percutaneous coronary intervention folllowing pre-hospital diagnosis of ST-elevation myocardial infarction. European Heart Journal 2006; 27: 1550-1557. [ Links ]
86. Mendoza C, Bhatt M, Virani S, et al. Management of failed thrombolysis after acute myocardial infarction: An overview of current treatment options. International Journal of Cardiology 2007; 114: 291-299. [ Links ]
87. Cantor W, Fitchett D, Borgundvaag B, et al. Routine Early Angioplasty after Fibrinolysis for Acute Myocardial Infarction. N Engl J Med 2009; 360: 2705-2718. [ Links ]
88. Davies C. Revascularization for cardiogenic shock. Q J Med 2001; 94: 57-67. [ Links ]
89. Wilansky S, Moreno C, Lester S. Complications of myocardial infarction. Crit Care Med 2007; 35: S348-S354. [ Links ]
90. Burkhart H, Dearani J. Surgical Management of Mechanical Complications of Acute Coronary Syndromes Causing Cardiogenic Shock. Contemporary Cardiology: Cardiogenic Shock: Diagnosis and Treatment. Cap 10 : 135-149. [ Links ]
91.Gowda R, Fox J, Khan I. Cardiogenic Shock: Basics and clinical considerations. International Journal of Cardiology 2008; 123: 221-228. [ Links ]
92. Overgaard C, Dzavík V. Inotropes and Vasopressors: Review of Physiology and Clinical Use in Cardiovascular Disease. Circulation 2008; 118: 1047-1056. [ Links ]
93. Asfar P, Hauser B, Radermacher P, et al. Catecholamines and Vasopressin During Critical Illness. Crit Care Clin 2006; 22: 131-149. [ Links ]
94. Lawson NW, Wallfisch HK. Cardiovascular Pharmacology: A new look at the pressors. Advances in Anesthesia. Edited by Stoelting RD, Barash PG, Gallagher TJ. Chicago, Year Book Medical Publishers, 1986, pp 195-270. [ Links ]
95. den Uli C, Lagrand W, Valk S, et al. Management of Cardiogenic Shock: Focus on Tissue Perfusion. Curr Probl Cardiol 2009; 34: 330-349. [ Links ]
96.Cooper B. Review and Update on Inotropes and Vasopressors. AACN Advanced Critical Care 2008; 19: 5-15 [ Links ]
97. Bayram M, De Luca L, Massie B, et al. Reassessment of Dobutamine, Dopamine, and Milrinone in the Management of Acute Heart Failure Syndromes. Am J Cardiol 2005; 96: 47G-58G. [ Links ]
98. Padayachee L. Levosimendan: The Inotrope of Choice in Cardiogenic Shock Secondary to Takotsubo Cardiomyopathy? Heart, Lung and Circulation 2007; 16: S65-S70. [ Links ]
99. Metzsch C, Liao Q, Steen S, et al. Levosimendan cardioprotection reduces the metabolic response during temporary regional coronary occlusion in an open chest pig model. Acta Anaesthesiol Scand 2007; 51: 86-93. [ Links ]
100. Toller W, Stranz C. Levosimendan, a New Inotropic and Vasodilator Agent. Anesthesiology 2006; 104: 556-569. [ Links ]

