










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Cardiología
Print version ISSN 1409-4142
Rev. costarric. cardiol vol.6 n.1 San José Jan. 2004
Introducción
El aire es una mezcla de gases entre los cuales se encuentran principalmente el nitrógeno (78,62%), el oxígeno (20,84%), el dióxido de carbono (0,04%) y el agua (0,50%).1
Sin embargo, es el oxígeno el que despierta más pasión, no sólo porque es indispensable para la vida al ser el aceptor final de la cadena respiratoria, sino también porque se ha vinculado en una serie de procesos patológicos denominados en conjunto estrés oxidativo.
Es por esta razón que en esta revisión se van a retomar los beneficios de los aspectos fisiológicos y fisiopatológicos del manejo de la cinética celular del oxígeno en pacientes gravemente enfermos, pero a la vez también se va a rescatar como este mismo gas se puede ver involucrado en el deterioro del metabolismo celular sistémico.
Fisiología del Transporte de Oxígeno
La difusión del oxígeno a los tejidos es posible gracias a una cascada de gradiente de presión, desde el aire ambiental hasta la mitocondria.2
Por ejemplo, a nivel del mar la presión barométrica es de 760 mmHg y la presión parcial de oxígeno (PO 2) a la inspiración es de 160 mmHg, considerando que el aire que respiramos contiene un 21% de oxígeno.
A su paso por las vías respiratorias, el aire se entibia y humedece; y de este modo, por la influencia de la presión de vapor de agua a nivel alveolar la PO 2 disminuye a un valor de 110 mmHg aproximadamente.
A continuación, por el efecto de la PCO 2 y de la difusión a través de la membrana alveolo capilar, la PO 2 en los capilares pulmonares es de 100 mmHg y al llegar a la aurícula izquierda se reduce a 95 mmHg a causa del cortocircuito anatómico. En la sangre que se transporta a los tejidos dicha presión es de 90 mmHg y en los capilares es de 40 mmHg. Se cree que la PO2 intersticial es de 10-20 mmHg, que a nivel de la membrana celular es de 10 mmHg y en la mitocondria oscila entre 1 y 5 mmHg.
Cuando el oxígeno difunde a través de la membrana alveolo capilar, el 97% se une a la hemoglobina y el 3% restante permanece disuelto en el plasma.1
La hemoglobina consiste en cuatro cadenas polipeptídicas y cuatro grupos hem; es un dímero de dímeros con dos cadenas de la familia alfa y dos cadenas de la familia beta. Las cuatro cadenas son mantenidas juntas por atracciones no covalentes. Cada cadena contiene un grupo hem que se une al oxígeno. Cualquier molécula de este tipo debe ser capaz de unir O2 , no permitirle que oxide ninguna otra sustancia (lo que reduciría el O2), y luego liberar el O2 en función de la demanda. Lo anterior se logra gracias a determinados metales de transición, en sus estados de oxidación más bajos, como por ejemplo, el Fe 2+ y el Cu 2+ que tienen una fuerte tendencia a unir oxígeno.
La razón fundamental de la existencia de proteínas que transportan oxígeno, además de los aspectos de solubilidad de dicho gas, es la protección de que el metal que une al oxígeno no sufra una oxidación irreversible, permitiéndose que se produzca el primer paso de una oxidación, o sea, la unión al oxígeno, pero se bloquea el paso final que es la oxidación completa.4
Una proteína transportadora de oxígeno ideal debería estar casi saturada a 100 mmHg e instaurada a aproximadamente 20-40 mmHg. (Figura 1)
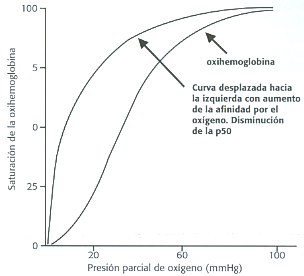
Entre las distintas cadenas laterales de los residuos de aminoácidos en la hemoglobina existen interacciones salinas, enlaces de hidrógeno e interacciones hidrofóbicas para estabilizar la determinada estructura cuaternaria. Estas interacciones son las que permiten cambios moleculares en la estructura de la proteína cuando por ejemplo ocurre un incremento de hidrogeniones, óxido nítrico, monóxido de carbono o cuando varía la concentración plasmática de 2,3 DPG y que por ende influyen en la curva de disociación de la oxihemoglobina. Es por esta razón que dicha curva posee una forma sigmoidea, o sea, que la pérdida de algunos oxígenos de la proteína facilita el que esta pierda más y viceversa, hecho que se logra por la intervención entre las distintas cadenas polipeptídicas de la hemoglobina. (Figura 1)
En el sentido inverso, cuando se produce CO 2 producto del metabolismo celular, este CO2 ingresa a los hematíes y reacciona con el agua y este proceso se aligera gracias a la enzima anhidrasa carbónica (AC), dando como resultados bicarbonato e hidrogeniones. Estos últimos iones reducen el pH de los eritrocitos, lo cual produce una reducción de la afi nidad de la Hb por el oxígeno, que a su vez permite una liberación aún más eficaz del oxígeno por parte de la hemoglobina. (Figura 2)
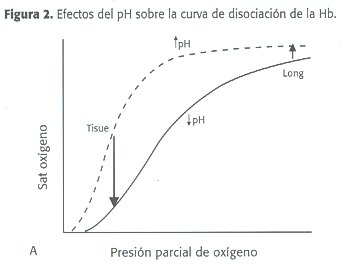
Lo anterior se ha denominado efecto Bohr y se describe de la siguiente manera:
De la fórmula anterior, se desprende que si ubicamos esta reacción en los pulmones, el exceso de oxígeno la desplazará hacia la izquierda, liberando H + . Estos H + reaccionan con el HCO-3 y forman CO 2 y agua como se explicó anteriormente, lo cual libera CO2 que posteriormente se expulsará por los pulmones.
Pero, ¿por qué si también se forman iones HCO3- el pH de los hematíes disminuye?
Parte de la respuesta está en que estos iones son intercambiados con iones Cloruro y se exportan fuera del hematíe; además parte del bicarbonato que permanece dentro del eritrocito reacciona con los grupos amino N-terminales de la hemoglobina para formar carbamatos, mediante una reacción denominada carbamación:
Como se aprecia, esta reacción introduce un grupo con carga negativa en el N-terminal de las cadenas, que estabiliza la formación de puentes salinos entre las cadenas ![]() y
y ![]() . El proceso ocurre en viceversa en los pulmones, expulsando el CO2 .4
. El proceso ocurre en viceversa en los pulmones, expulsando el CO2 .4
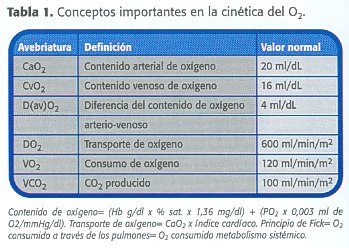
La cantidad de oxígeno transportado a los tejidos periféricos es el producto del contenido de oxígeno en sangre arterial por el gasto cardíaco (GC), es decir, de 1000 ml/min o de 600 ml/ min/m 2 aproximadamente.5
A su vez, el contenido de oxígeno es la suma del oxígeno unido a la hemoglobina más el oxígeno disuelto en sangre. 4
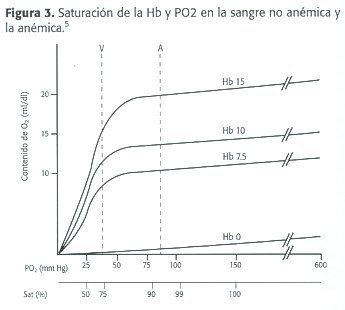
Para realizar este cálculo, es necesario recordar que cada gramo de Hb se une a 1.36 ml de oxígeno, por lo tanto, si la Hb normal es de 15 g/dl y esta se encuentra saturada al 100%, la cantidad de oxígeno unido a la hemoglobina es de 20.4 mg/dl. Por otro lado, el coeficiente de solubilidad del oxígeno es de 0.0031 ml/mm Hg/dL, y por consiguiente, la cantidad de oxígeno disuelto en 1 dL de sangre con una PO2 de 100 mm Hg es de 0.3 ml, por lo que el contenido de oxígeno en la sangre arterial es de 20.7 ml/dL. (Tabla 1)
Utilizando el mismo cálculo, el contenido para la sangre venosa es de aproximadamente 16 ml/dL, por lo que la diferencia arterio-venosa de oxígeno (DavO2 ) es de 4.7 ml/dL. Un concepto importante a destacar es que la PO2 y la saturación son las mismas para la sangre anémica arterial y venosa, incluso cuando existe una intensa disminución del contenido del oxígeno. (Figura 3)
Al analizar el transporte de oxígeno ( DO 2 ) y el consumo (VO2 ) calculado vs. el medido, hay que mencionar que hay diferencias importantes. Por ejemplo, el VO2 calculado puede variar en un 15% mientras la medida en un 5%.6,7
Normalmente, existe una relación entre el DO 2 y el VO2 de 5:1 y ante cualquier variación de componentes del transporte de oxígeno (Hb, sat, PO2 , índice cardíaco), los otros tienden a modificarse para de esta forma compensar el cambio en dicha variable y mantener constante el DO2 . Por ejemplo, en un paciente anémico agudo, aumenta el GC hasta que el DO2 se restablece. Cuando la anemia es crónica, no sólo aumenta el GC, sino también el número de hematíes, como consecuencia de la estimulación de la eritropoyetina.
Sin embargo, cuando es el gasto cardíaco el que ha alterado la relación DO2 /VO2 de forma aguda, el parámetro que se autorregula para mantener dicha relación es el VO 2, es decir, se extrae una cantidad relativamente mayor de oxígeno de la sangre circulante, con lo cual se incrementa la DavO2 .
Un cambio primario del DO2 no va seguido de ningún cambio del VO2 .
Es evidente que el VO2 no puede superar el DO2 y si el DO2 es menor que el VO 2 , el VO 2 llegaría a ser dependiente del suministro. En teoría esto ocurriría cuando el cociente fuera inferior a 1:1. No obstante, se produce una dependencia del suministro de oxígeno cuando el DO2 disminuye por debajo del doble del VO2 (2:1). (Figura 4)
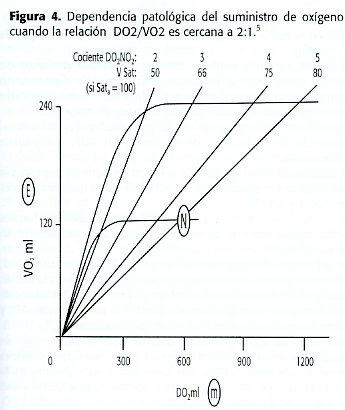
La cantidad de oxígeno extraído del DO 2 es un 20% y el 80% restante en realidad se encuentra presente en la sangre venosa de retorno al corazón; por lo tanto, la saturación de sangre venosa (SvO2 ) es del 80%.
De lo anterior se deduce que cuando la SvO 2 es del 80%, el cociente DO2 /VO2 se encuentra en su estado 5:1. Una SvO2 del 50% corresponde a una relación 2:1 (siempre que la sangre arterial esté saturada al 100%). Ahora, si la SaO2 es del 80% y la SvO2 es del 64%, entonces el cociente es de 5:1. La SvO2 puede estar elevada en tejidos que han estado hipoperfundidos, como por ej. durante la circulación extracorpórea en la cirugía de corazón.
Cabe resaltar que en ciertos procesos clínicos, como en la sepsis, la curva DO2 /VO2 se desvía hacia la derecha y el cociente crítico DO2 /VO2 podrían acercarse a 3:1 más que a 2:1, lo cual podría deberse a problemas de difusión desde los capilares a las mitocondrias o a las anomalías en cadena respiratoria. (ver Figura 4)
LAS MITOCONDRIAS Y EL CONSUMO DE OXÍGENO
Las mitocondrias se encuentran en casi todas las células (a excepción de los hematíes). Su número varía según el tipo celular, por ejemplo cada hepatocito posee de 1.022 a 2.000 mitocondrias, que miden 3 milimicras de largo aproximadamente.
Las mitocondrias poseen dos membranas, una externa y otra interna que dan lugar a los compartimientos intermembranosos y a la matriz mitocondrial.
Es en la matriz mitocondrial y en la membrana interna en donde se desarrollan la mayoría de actividades relacionadas a la cadena respiratoria. 8,9
Directamente relacionado con estos procesos aerobios intramitondriales, se encuentra el consumo de oxígeno, es decir, el volumen de oxígeno consumido por minuto (VO2 ) y se aprecia mejor observando de una manera más detallada la cadena respiratoria. (Figura 5)
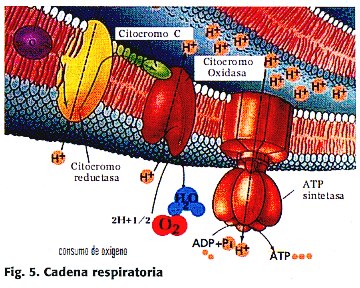
No obstante, el consumo de oxígeno por los tejidos es un poco mayor que la producción aeróbica de ATP. La explicación de lo anterior, puede ser la utilización de dicho gas en otros procesos oxidativos celulares, que según se calcula consumen el 2% del VO2 , como por ejemplo, en la generación de metabolitos reactivos del oxígeno.10, 11
El VO2 en reposo está en función de la respiración celular, y esta a su vez, depende del ambiente metabólico de los diferentes órganos, regido en especial por los niveles hormonales. 12
El metabolismo que se produce en diferentes órganos ocurre a distintas velocidades que dependen de la masa celular y de la actividad celular, de modo que el VO2 sistémico se ve afectado por los cambios del flujo sanguíneo regional.
El VO2 se puede calcular de diversa formas, pero en general estas se basan en el principio de Fick que establece que en un estado de equilibrio, la cantidad de O2 consumido en el proceso del metabolismo sistémico es exactamente igual a la cantidad de oxígeno captado en los capilares pulmonares por medio de la vía aérea. A su vez, la eficiencia de la captación de oxígeno a través de los pulmones está controlado por la relación perfusión-ventilación alveolar.
La respiración celular se defi ne como la transferencia ordenada de vías metabólicas electrónicas, de compuestos orgánicos al oxígeno. Este proceso genera fosfatos de alta energía en la forma de ATP para aportar energía libre necesaria por las células de los tejidos. 13
El ATP es hidrolizado por las ATPasas para generar energía necesaria para las funciones celulares y para conservar la permeabilidad de la membrana de la célula.14
La hidrólisis del ATP hace que se produzcan ADP, fosfato inorgánico (Pi) e iones hidrógeno.
La respiración celular y por ende la fosforilación oxidativa están controlados a nivel celular por varias señales; dentro de las cuales el potencial de fosforilación (PF) es una de las más importantes.
Dicho potencial se define como
Es decir, cuando el PF es alto (o sea que la cantidad de ATP es elevada y por ende la célula se encuentra con un balance energético a su favor) disminuye el metabolismo aeróbico y por ende el consumo de oxígeno.15
Por otro lado, si se acelera la hidrólisis de ATP, como sucede durante el ejercicio, aumentan la (ADP) y (Pi) y por lo tanto disminuye el PF y es así como aumenta la respiración celular, el consumo de oxígeno y el flujo sanguíneo a dichos tejidos. (En este último caso hay un aporte de sustrato en forma de ADP en la fosforilación oxidativa, lo que hace que esta pueda aumentar).
El (ADP) y el (Pi) también aumentan durante procesos de hipoxia, pero ante la limitación del aporte de oxígeno, no aumenta el consumo de este gas y dicho consumo disminuye conforme la privación del gas mencionado es mayor. Otro factor determinante de la producción de ATP es el contenido de nucleótidos de adenina (ATP, ADP, AMP) en la mitocondria.
Al acumularse en el citosol, ADP y Pi son llevados de regreso a la mitocondria por translocasas de ADP/ATP y por transportadores de Pi/OH y ATP/Pi.
En condiciones normóxicas, la distribución de los fondos mencionados entre el citosol y la mitocondria se encuentran en equilibrio.16
Es así como diversas moléculas que influyen sobre le VO2 actúan a nivel mitocondrial, por ejemplo, las hormonas tiroideas actúan directamente sobre el complejo IV o Cit. C. Oxidasa, y de esta forma eliminan la inhibición alostérica por parte del ATP y como consecuencia aumentan la respiración celular. 17
Otro ejemplo es el aumento del ATP, que produce que se desate una señal de transducción vía proteínas G que conlleva aumentos de AMPc y PKA, con fosforilación subsiguiente de la Cit. C. Oxidasa e inhibición de este complejo, que como ya se ha explicado disminuirá el VO2 . 18
Por otro lado, el aumento de la concentración de Ca ++ mitocondrial activa el complejo IV al desfosforilarlo y por ende aumenta también el VO2 .
Papel del oxido nítrico a nivel mitocondrial y su relación con el consumo de oxígeno
El óxido nítrico (NO) representa una de las 10 moléculas de menor tamaño halladas en la naturaleza y está compuesto por sólo un átomo de nitrógeno y un solo átomo de oxígeno.
La molécula de NO contiene un número impar de electrones (cinco electrones del nitrógeno más seis electrones del oxígeno) lo que implica que existe un electrón desapareado y que confiere al NO la propiedad de molécula reactiva.
El NO es una molécula sumamente lipofílica y que por lo tanto atraviesa fácilmente las barreras representadas por las membranas lipoproteicas. Presenta una vida media de apenas 3-5 seg., debido a su reactividad espontánea con el oxígeno molecular. Se sintetiza a partir de la L-arginina (un aminoácido básico) por la acción de la NO sintetasa, que también cataliza una segunda reacción para dar como resultado la L-citrulina. Presenta una afinidad por la hemoglobina 200.000 veces superior a la del oxígeno y 1.000 veces mayor que la del monóxido de carbono.18 (Figura 6)
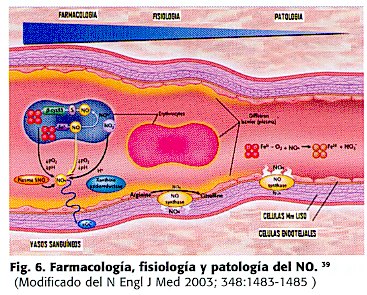
La actividad fisiológica de NO finaliza rápidamente después de su oxidación en presencia de oxígeno para formar nitrito (NO2) y nitrato (NO3).
Esta molécula actúa a nivel mitocondrial en situaciones de estrés celular en una forma compensadora, es decir, tratando de evitar la muerte celular.19,20,21.
Sin embargo, pareciera que este mismo metabolito es el que también puede provocar que una célula que ha dejado de ser viable se conduzca al proceso conocido como apoptosis.
La apoptosis (o muerte celular programada) es en última instancia un mecanismo de defensa celular ante una agresión, tanto endógena como exógena y que pone en peligro el funcionamiento celular. También se ha relacionado este proceso de apoptosis en una amplia gama de patologías, por ejemplo, la aterosclerosis, la insuficiencia cardíaca, eventos isquémicos cardíacos y cerebrales, rechazo de trasplantes cardíacos y otros. 22
El Óxido Nítrico inhibe la citocromo oxidasa reversiblemente, al competir por el oxígeno por los sitios A3 y CuB reducidos, en otras palabras, aumenta la Km de la enzima por el oxígeno.
Se sabe que una disminución en el aporte de oxígeno conlleva a una generalización de NO.
El NO entonces produciría una caída en la actividad de los complejos I, III, IV, que bombean protones desde la luz de la matriz mitocondrial hasta el espacio intermembranoso. Existe un intercambiador activo secundario que se denomina translocador de nucleótidos de adenina (ANT), que normalmente saca ATP al espacio intermembrana mitocondrial (EIM) en intercambio por ADP (ATP tiene una carga negativa de más que el ADP). Este intercambiador también se vería frenado, ya que el "motor" de todo lo anterior es la Cit. Oxidasa, (la cual reduce al oxígeno) y que en este caso está inhibida por el NO.20
Otra situación que ocurre en caso de hipoxia, además de todo lo anterior, es que la ATP sintetasa o complejo V, en lugar de bombear los protones al interior de la matriz mitocondrial, el proceso se revierte y se bombean protones al EIM, como una medida para tratar de restablecer el gradiente electroquímico. Por lo tanto, en un grado de "hipoxia leve" para la célula, estos mecanismos compensadores provocan que el potencial de membrana mitocondrial se restablezca y que la célula pueda evitar así la apoptosis.
No obstante, el estado reducido de la cadena respiratoria favorece la generación de iones superóxido, que en este caso son convertidos a peróxido de hidrógeno por la peróxido dismutasa (SOD). Ahora bien, conforme la hipoxia prosigue, esta inhibición de la respiración incrementa la producción de radicales libres, se agota el pool de glutatión y la formación de peroxinitritos aumenta.22
Esto último favorece la inducción del poro de Transicional de Permeabilidad (PTP) en la membrana mitocondrial interna (MMI), que conlleva que la MMI se vuelva permeable a los H+ y se provoque un colapso en la célula, con la liberación citocromo C e inducción de un complejo (apoptosoma) que induce la activación de la cascada apoptótica. 23
De lo anterior se desprende que durante la hipoxia el NO juega un papel protector inicialmente, disminuyendo parcialmente el consumo de oxígeno celular. Es así como, en la disminución de la PO 2 celular, la producción de ATP es menor que las necesidades de la célula, y se acumula AMP citosólico, ello a su vez estimula la glicólisis con la estimulación del lactato, al estimularse la enzima lactato deshidrogenasa que convierte el piruvato en lactato y oxida el NADH + H+ y es a este incremento de la rapidez de la glucólisis con la hipoxia lo que se conoce como Efecto Pasteur.
Otra fuente de ATP como mecanismos compensatorios son las reacciones de la cinasa de creatina, en la cual se utiliza fosfocreatina para fosforilar ADP. Sin embargo, no tiene aporte importante durante estados de hipoxia, ya que la fosfocreatina disminuye importantemente durante estos eventos. La CPK, sin embargo, acumula la concentración de ADP citosólico, lo cual a su vez, estimula la adenilato quinasa la cual intenta conservar la relación, ![]() al fosforilar un ADP a partir de otro ADP. Esto conllevaría a un aumento de (AMP) citosólico, el cual puede tomar dos vías: o se desamina y produce monofosfato de inosina (IMP), con la formación de amoníaco, o es convertido por la enzima 5’ nucleotidasa a adenosina que es un vasodilatador potente. 9,16
al fosforilar un ADP a partir de otro ADP. Esto conllevaría a un aumento de (AMP) citosólico, el cual puede tomar dos vías: o se desamina y produce monofosfato de inosina (IMP), con la formación de amoníaco, o es convertido por la enzima 5’ nucleotidasa a adenosina que es un vasodilatador potente. 9,16
También la cascada metabólica implica a la enzima xantino oxidasa, que puede producir radicales libres y peroxidación de membranas; formación de metabolitos del ácido araquidónico y mayor alteración de la microvasculatura corporal.24
De ahí es que se ha intentado administrar inhibidores de la xantino oxidasa y vitamina E durante eventos de reperfusión, donde se sabe que ocurre una producción acelerada de radicales libres y que pueden contribuir incluso a un deterioro de la función celular más que a su viabilidad. 24
NOTA : El artículo se divide en dos partes: un primer capítulo que consiste en la fi siología de la cinética del oxígeno y una segunda parte que abarca la correlación clínica del tema.
Referencias
1. Guyton AC, Hall JE. Textbook of Medical Physiology. 9th ed.Philadelphia,W. B. Saunders, 1996. [ Links ]
2. Ganong WF. Review of Medical Physiology.19th ed. California, Appleton and Lange, 1999. [ Links ]
3. Dickerson, R.E. e I. Geis: Hemoglobin: Structure, Function, Evolution and Pathology. Benjamin/Cummings, Menlo Park, California, 1983. [ Links ]
4. Mathews, C. K., van Holde, K.E., Ahern K.G. Biochemistry. 3rd ed. Addison Wesley, California, 2002. [ Links ]
5. Bartlett RH. Critical Care Physiology. Masson-Little, Brown, 1996. [ Links ]
6. Dantzker DR, Foresman B, and Gutierrez G: Oxygen supply and utilization relationships: A reevaluation. Am Rev Respir Dis 143: 675-679, 1991.
7. Schneeweiss B, Druml W, Graninger W, et al. Assessment of oxygen-consumption by use of reverse Fick principle and indirect calorimetry in critically ill patients.Clin Nutr 1989;8:89-9 [ Links ]
8. De Robertis EMF, Biología Celular y Molecular. 12ª ed., 4ª reimp. Buenos Aires: El Ateneo, 1998 [ Links ]
9. Cooper GM. The Cell: A Molecular Approach. 2nd ed. Boston, Sinauer Associates, 2002. [ Links ]
10. Bartlett RH, Dechert RE. Oxygen Kinetics: pitfalls in clinical research. J Crit Care 1990;5:77-80 [ Links ]
11. Grisham MB and McCord JM: Chemistry and cytotoxicity of reactive oxygen metabolites. In Taylor AE, Matalon S, and Ward P,.: Physiology of oxygen radicals. Bethesda, MD, American Physiological Society, 1986, pp 1-18.
12. Bartlett RH. Critical Care. In: Greenfield L, ed. Surgery: scientific principles and practice. Philadelphia: Lippincott, 1992: 195-222. [ Links ]
13. Di Mauro S, Schon EA. Mitochondrial Respiratory Chain Disease. N Engl J Med 2003; 348:2656-2668. [ Links ]
14. Harold FM: The vital force: A study of bioenergetics. New York, Freeman, 1986,pp 28-56 [ Links ]
15. Saraste M: Oxidative phosphorylation at the fin de siecle. Science 283:1488-1493. [ Links ]
16. Gutiérrez G: Cellular energy metabolism during hypoxia. Crit Care Med 1991; 19:619. [ Links ]
17. Ludwing B, Bender E, et al. Cytochromec Oxidase and the Regulation of Oxidative Phosphorylation.Chembiochem 2001;2:392-403. [ Links ]
18. Berne R.M. and Levy M.N.: Principles of Physiology.4th ed. Mosby .1998 [ Links ]
19. Matheis G, Sherman MP, Bucberg GD, et al: Role of L-arginine-nitric oxide pathway in myocardial reoxygenation injury. Am J Physiol 1992; 262:H616-H620
20. Moncada S, Erusalimsky JD. Does nitric oxide modulate mitochondrial energy generation and apoptosis?. Nature 2002; 3:214-220. [ Links ]
21. Gianetti J, Del Sarto P, et al.: Supplemental nitric oxide and its effect on myocardial injury and function in patients undergoing cardiac surgery with extracorporeal circulation.J Thoracic Cardiovasc. Surg 2004;127:44-50 [ Links ]
22. Palmer, L.A., Gaston, B. and Johns, R.A. Normoxic stabilization of hipoxia inducible factor 1 expression and activity: redox dependent effect of nitrogen oxide. Mol Pharmacol. 58, 1197-1203
23. Narula J, Dixit VM, Miller LW. Apoptosis in cardiovascular disease. Cardiology Clin 2001; 19:95-150 [ Links ]
24. Bontemps F, Van den Berghe G, and Hers HG: Pathways of adenine nucleotide catabolism in erythrocytes. J Clin Invest 77:824-830,1986. [ Links ]
* Asistente en Cirugía Cardiovascular, Hospital México, C.C.S.S., San José, Costa Rica.
** Residente en Circulación Extracorpórea, Hospital México, C.C.S.S., San José, Costa Rica.
*** Médico General.
**** Asistente en Cardiología y Circulación Extracorpórea, Hospital México, C.C.S.S., San José, Costa Rica.

