










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Salud Pública
Print version ISSN 1409-1429
Rev. costarric. salud pública vol.21 n.2 San José Jul./Dec. 2012
Ensayo
Diagnóstico y tratamiento de retinoblastoma
Diagnosis and treatment of retinoblastoma
Meiting Nie
*Dirección para correspondencia:
El retinoblastoma es el tumor maligno más frecuente en la infancia, sin embargo sigue siendo un tumor raro en niños. Se asocia a una inactivación de ambos alelos del gen supresor tumoral. Pueden aparecer esporádicamente o pueden ser familiares y existen varios patrones de diseminación tumoral que pueden variar el pronóstico tanto visual como del globo ocular. El signo más frecuente es la leucocoria seguido por estrabismo, con una edad promedio de presentación a los 2 años en casos unilaterales y durante del primer año de vida en los tumores bilaterales. La detección temprana y tratamiento adecuado permite una mayor sobrevida en el niño.
Palabras Claves: Retinoblastoma, Neoplasia Maligna, Diagnóstico (fuente: DeCS, BIREME).
Abstract
Retinoblastoma is the most common malignant tumor in childhood; however it is ar are tumor in children. It is associated with inactivation of both alleles of the tumor suppressor gene. It may occur sporadically or may be family related; there are various patterns of tumor dissemination that can vary the visual and eye prognosis. The most common sign is leukocoria followed by strabismus, with an average age of onset at 2 years in unilateral cases and during the first year of life in bilateral tumors. Early detection and appropriate treatment can prolong survival in children.
KeyWord: Retinoblastoma, Malignant Neoplasm, Diagnosis. (Source: MeSH, NLM).
Aproximadamente el 60 %tienen un patrón esporádico con tendencia a ser unilaterales y solitarios, el resto son de patrón hereditario, comúnmente bilaterales y multicéntricos.
La mayoría se presentan por una mutación nueva en las células germinales y sólo un 10 % son de casos familiares con un defecto recesivo a nivel celular (2). Un
Patrones de diseminación. El tumor puede ser de crecimiento exofítico hacia el espacio subretiniano o endofítico hacia el vítreo, de modo que pueden producir invasión del nervio óptico con la consecuente diseminación al cerebro a través del espacio subaracnoideo, a los tejidos orbitarios por las venas emisarias y nervios en la esclerótica, y también pueden llegar a realizar metástasis (4).
Cuadro Clínico. La presentación más frecuentemente observada es la leucocoria (reflejo pupilar blanco), seguido por estrabismo, y ocasionalmente se puede encontrar una reducción de la agudeza visual, hiperemia conjuntival, exoftalmos debido a una inflamación orbitaria que puede simular una celulitis orbitaria o preseptal (5).
Los casos bilaterales presentan una edad promedio en el primer año de vida y los unilaterales hacia los dos años de vida. Los signos clínicos pueden variar según el patrón crecimiento y las dimensiones en el momento del diagnóstico, por lo tanto cabe recordar que es necesario realizar tanto oftalmoscopia directa como indirecta con indentación escleral en los ambos ojos bien dilatados para no subdiagnosticar retinoblastomas en la retina preecuatorial (6).
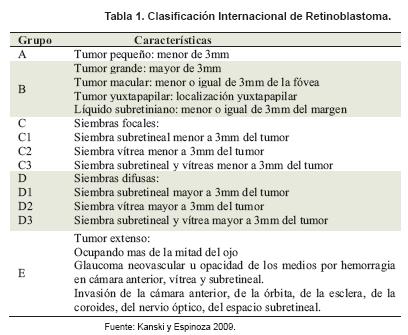
Clasificación Internacional de Retinoblastoma. En 1960 se creó la clasificación Reese–Ellsworth determinado en cinco grupos, basado en el estadio del tumor intraocular y la probabilidad de preservar el ojo luego del tratamiento con radioterapia con haz externo. A partir del 2003 surge la clasificación internacional del retinoblastoma (6-7). Tabla 1
Diagnóstico Diferencial. Otras causas de leucocoria son: retinopatía del prematuro, cataratas de diversas causas, persistencia del vítreo primario hiperplásico, enfermedad de Coats, uveítis por toxocara, uveítis crónica (8). Por consiguiente es fundamental realizar una ecografía del globo ocular y tomografía axial computarizada, en donde puede observarse calcio en el retinoblastoma (9).
Metástasis. Menos del 10 % de los pacientes presenta metástasis y los lugares más frecuentes son huesos del cráneo y de la órbita, a través de la diseminación hematógena, siendo ésta la vía más común seguida porinvasiónal nervio óptico. Otros sitios afectados son: cerebro, huesos largos, ganglios linfáticos, hígado, riñón, gónadas, páncreas y pulmones. El pronóstico es reservado y comúnmente fallecen a los seis meses (10).
Diagnóstico. Es importante realizar una historia clínica completa, teniendo en cuenta que los pacientes con antecedentes familiares de retinoblastoma deben ser evaluados de forma periódica desde el nacimiento. Otros estudios son: una revisión exhaustiva de ambos ojos externamente, oftalmoscopia indirecta, la toma de presión intraocular, radiografía ocular, la ecografía es una herramienta útil para evaluar tanto el tamaño del tumor como las calcificaciones, la tomografía computarizada también detecta calcificaciones (11).
Tratamiento. El primer objetivo del tratamiento del retinoblastoma es mejorar la sobrevida del paciente y en segundo lugar salvar el globo ocular con conservación de la mejor agudeza visual posible, y es de manejo multidisciplinario por: oftalmólogo, pediatra, oncólogo pediatra, patólogo, genetista, trabajo social, enfermería, y otros que juegan un papel importante en la cura de la enfermedad. El manejo varía en cada niño, es fundamental tener en cuenta los factores como el tamaño, la localización del tumor, presencia de metástasis y la producción de neoplasias secundarias.
La radioterapia con haz externo y la enucleación fueron técnicas muy usadas, sin embargo en la actualidad existen diversos tratamientos conservadores utilizados para tumores pequeños y en estadios precoces como: fotocoagulación con láser, crioterapia, termoterapia transpupilar, braquiterapia y quimioterapia focalizada. La quimioterapia sistémica es útil para reducir el tamaño del tumor pero se asocia con muchos efectos secundarios y cánceres secundarios como leucemia, por estas razones se introdujo la quimioterapia focalizada subconjuntival y la quimioterapia superselectiva el cual se introduce el agente dentro de la arteria oftálmica. Los tratamientos anteriores permiten preservar el globo ocular. La radioterapia, la enucleación y excenteración se reservan para estadios más avanzados.Actualmente han introducido nuevas terapias como agentes antiangiogénicos ( acetato de anecortave) e inhibidores glucolíticos ( 2desoxi-D-glucosa) muy valiosas en el tratamiento del retinoblastoma (12).
Terapia Focal. La braquiterapia consiste en la aplicación de yodo 125, oro o rutenio 106, la cual está indicada en tumores con un tamaño menor de 16mm de base y menor de 8mm en grosor, también se puede usar como tratamiento primario o en casos de fallo a la radioterapia, pero con la desventaja de producir cataratas, retinopatía y neuropatía óptico como efectos secundarios. La termoterapia utiliza el calor y se aplica sobre el retinoblastoma usualmente con un tamaño menor o iguala3mmdediámetro,siemprequenoexistasiembra vítrea o subretineal. Existe una modalidad llamada quimiotermoterapia que combina la quimioterapia con la termoterapia utilizados para tratar tumores de mayor tamaño o con presencia de siembras.
La fotocoagulación con láser produce coagulación de la vasculatura tumoral, indicado para retinoblastomas posteriores de pequeño diámetro, y los efectos adversos incluyen desprendimiento de retina, oclusión vascular retiniana, tracción retiniana y fibrosis.
La crioterapia funciona causando trombosis e infarto por daño al endotelio vascular del cáncer, aplicado para masas periféricas pequeñas, y uno de sus efectos dañinos son edema conjuntival, desprendimiento de retina y hemorragia vítrea (13).
Quimioterapia. La quimioterapia con carboplatina, etopósido y vincristina con o sin ciclosporina por vía intravenosa es el más usado para el tratamiento primario del retinoblastoma intraocular, con el objetivo de reducción del tamaño para posteriormente ser tratado con terapia focal, gracias a esta combinación cada vez se usa menos la enucleación y la radioterapia, así disminuir las complicaciones que producen ambas (14).
Radioterapia con Haz Externo. Se administra en casos avanzados cuando existen siembras vítreas difusas, a pesar que es un tumor radiosensible existe la posibilidad de recurrencia dentro de los primeros 4 años posterior a la radiación. En lo posible se trata de evitarlo debido al riesgo de inducir daño de la retina, del nervio óptico, cristalino y una segunda neoplasia maligna en pacientes con retinoblastoma hereditario, asociados más frecuentemente en niños menores de 12 meses de edad.
Enucleación. La enucleación está indicado en enfermedad avanzada donde existe pobre visión útil o posibilidad de invasión hacia el nervio óptico, coroides y órbita (15).
Tratamiento con inhibidores glucolíticos (2- desoxi -Dglucosa). Generalmente existen áreas hipóxicas en tumores sólidos que presentan proliferación celular lenta que dependen de una glicólisis anaerobia para producir ATP (adenosín trifosfato) y sobrevivir. La quimioterapia y la radioterapia actúan sobre células con división rápida, por lo tanto el tejido hipóxico de lento crecimiento es más difícil de atacar por ambas. Debido a lo anterior se creó una terapia (2 -desoxi-D- glucosa) que es capaz de inhibir la glicólisis produciendo agotamiento de las reservas de ATP en las células cancerigenas y así la muerte celular. Los inhibidores glucolíticos son utilizados como coadyuvantes junto a la quimioterapia y han demostrado tener una mayor eficacia terapéutica. In Vitro han demostrado que el 2 -desoxi-D-glucosa rápidamente induce la muerte de células hipóxicas dentro de las 24 horas, bloqueando la glicólisis mediante la inhibición de hexoquinasa, permitiendo muerte selectiva de células tumorales hipóxicas y no las células normales aerobias (16).
Conclusiones
El retinoblastoma es un tumor que sigue siendo un desafío tanto a nivel diagnóstico como a nivel de tratamiento. Actualmente existen muchas terapias que han demostrado ser eficientes, pero un factor muy importante es la detección precoz, que junto con un manejo multidisciplinario adecuado mejora la sobrevida del niño. Por consiguiente es fundamental tener conocimientos sobre el tumor y educar tanto a la población médica como a los familiares para su detección.
Agradeciemientos
A mis padres que siempre me apoyaron durante toda mi carrera, al Ing. Federico Solano Rojas por el apoyo incondicional y a todos los que me mantienen con fe para seguir luchando por mis objetivos.
Referencias
1. Spalton D, Hitchings R, Hunter P, Tan J, Harry J. Atlas de Oftalmología Clínica. 3a Ed. España: Elsevier; 2006. [ Links ]
2. Cibis G, Beaver H, Johns K, Kaushal S, Tsai J, Beretska J. Fundamentos y Principios de Oftalmología. 2007-2008. España: Elsevier; 2008. [ Links ]
3. Rodríguez M, Prado M, Salcedo M. Perspectivas en la genómica del retinoblastoma: Implicaciones del gen supresor de tumor RB1. Revista de Investigación Clínica 2005; 57(4): 572-581. [ Links ]
4. Vaughan D, Asbury T, Riordan-Eva P. Oftalmología General. 11a Ed. México: El Manual Moderno; 1997: 237. [ Links ]
5. Trincado A, López J, González M, Villaseca E, Roizen A, Manieu D et al. Retinoblastoma en Pediatría, experiencia en un hospital pediátrico. Revista Chilena de Pediatría 2008; 79 (6): 614-622. [ Links ]
6. Kanski J. Oftalmología Clínica. 6a Ed. España: Elsevier; 2009: 538. [ Links ]
7. Espinoza M. Retinoblastoma. Revista Médica de Costa Rica y Centroamérica 2011; LXVIII (596):23-27. [ Links ]
8. Manzitti J, Mansilla M. Descripción del caso presentado en el número anterior: Retinoblastoma. Archivo Argentino de Pediatría 2010; 108(3):255 -257. [ Links ]
9. Escanio M, Murillo L. Retinoblastoma. Revista Mexicana de Oftalmología 2002; 76(6): 243 – 245. [ Links ]
10. Alvarado B, Campos L, Villavicencio A. Características Clínicas y Metastásicas en Retinoblastoma. Revista Médica del Instituto Mexicano del Seguro Social 2009; 47(2):151-156. [ Links ]
11. Martínez P, Zorrilla H. Retinoblastoma. Revista Médica Dominicana 2000; 61(3):241. [ Links ]
12. Villamil J, Quintero L, Serrano R, Moreno I. Consideraciones Cínicas, Diagnósticas y de Tratamiento en Retinoblastoma. MedUNAB 2012; 14(3):180-187. [ Links ]
13. Chintagumpala M, Chevez P, Paysse E, Plon S, Hurwitz R. Retinoblastoma: Review of Current Management. The Oncologist 2007; 12:1237 – 1246. [ Links ]
14. Kiss S, Leiderman Y, Mukai S. Diagnosis, Classification, and Treatment of Retinoblastoma. International Ophthalmology Clinics 2008; 48(2):135 – 147. [ Links ]
15. Shields C, Shields J. Diagnosis and Management of Retinoblastoma. Cancer Control 2004; 11(5): 317-325. [ Links ]
16. Boutrid H, Jockovich M, Murray T, Piña Y, Feuer W, Lampidis T et al. Targeting Hypoxia, a Novel Treatment for Advanced Retinoblastoma. Investigative Ophthalmology & Visual Science 2008; 49 (7): 2799 – 2804. [ Links ]
*Correspondencia a: Licenciatura en Medicina y Cirugía, Doctora en Medicina y Cirugía, Meiting477@gmail.com


{kind=link}