











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Los traumatismos cráneo encefálicos (TCE) son una de las primeras causas de mortalidad y discapacidad en los países desarrollados, sobre todo en la población menor de 45 años, debido, en parte, al aumento del uso de los vehículos a motor. De hecho, aproximadamente el 25% de las muertes violentas se deben a esta casusa. (1,2). Por otro lado también es importante aproximarnos al diagnóstico y al pronóstico de los traumatismos cráneo encefálicos por la relación que existe entre el trauma de intensidad moderada y las secuelas a largo plazo, que llegan a incluir enfermedades degenerativas como el Alzheimer y el Parkinson. (3,4,5,6).
Actualmente, la mayoría de los estudios a este respecto son de carácter experimental en animales o son series clínicas con una perspectiva clínica y pronóstica, en especial los que versan sobre el precursor del beta amiloide (β APP), la proteína glial fibrilar acida (PGFA), y la proteína SB100 (7,8). También existen estudios retrospectivos sobre casos necrópsicos que ponen en relación alteraciones del comportamiento con otros marcadores como la proteína Tau y la apolipoproteína E (APOE) (9,10) o también el mismo β-APP y su relación con la encefalopatía postraumática (11), pero todos ellos tienen más relevancia clínica que forense.
Las técnicas argénticas aún resultan útiles a día de hoy porque pueden mostrar como cambian las sinapsis tras un traumatismo (12), pero las técnicas inmunohistoquímicas son más sensibles y específicas, ya que nos permite observar la respuesta de determinada estirpe celular y es una técnica asequible en cualquier laboratorio (13). Entre ellas destaca, el β-APP que revela cambios bioquímicos en la neurona tras sufrir fenómenos de aceleración-desaceleración y torsión que alteran la permeabilidad de la membrana celular.
El β-APP es una glicoproteína transmembrana de la neurona. El β-APP parece intervenir en la adhesión y promover el crecimiento celular, además de presentar funciones autocrinas y actuar como sitio de unión para sustancias como la heparina. Es transportado por transporte axonal rápido anterógrado y se acumula tras la alteración del citoesqueleto axonal (14,15,16). En traumatismos de baja intensidad se ha demostrado en el soma neuronal y en dendritas distróficas de la zona contundida una vez que se ha producido la lesión axonal. Progresivamente, cuando su presencia en neuronas comienza a disminuir, el β-APP se ha demostrado, aunque en menor cantidad, en la microglía /macrófagos (con doble inmunofluorescencia con CD11b y beta-APP), si bien, dicha positividad se ha relacionado con la fagocitosis de los axones dañados (15).
Su acumulación en las neuronas no es específica del traumatismo craneoencefálico, sino que aparece en el daño axonal de distintas causas como hipoxia, alteraciones metabólicas y todas aquellas circunstancias en las que exista edema y/o aumento de la presión intracraneal. Es decir, constituye una respuesta del tejido cerebral a cualquier agente de estrés de cierta entidad. El acúmulo de β-APP se produce por la alteración del citoesqueleto por la proteólisis, junto con la alteración de las quinasas y las fosfatasas, aumenta la permeabilidad de la membrana; ésta altera el flujo de calcio y la actividad de la calmodulina y altera el transporte axonal debido a la desestructuración y colapso del citoesqueleto por la compactación de los neurofilamentos, la pérdida de los microtúbulos y la fragmentación de la espectrina. Como consecuencia, los materiales transportados se acumulan en lugares de máximo daño axonal y los axones sufren una axonotomía secundaria. (17,18).
Por otro lado, los estudios de Staal y cols (19), demuestran el paralelismo entre el acúmulo de calcio en la célula y la aparición del -APP. Se cree que la acumulación conjunta de BACE (b-secretase), PS-1 (presenilin-1), caspasa 3 activada y CCA (caspase-mediated cleavage of APP) con β-APP en los axones dañados en daño axonal difuso, es debida a un único mecanismo de producción de beta amiloide, secundario a la alteración de la permeabilidad de la membrana que daría lugar a su acumulación patológica (20).
De hecho, actualmente se está poniendo en valor la relación de la presencia de esta proteína con patologías neurodegenerativas tales como la Enfermedad de Alzheimer. Las alteraciones vasculares y la hipoxia secundaria a la inflamación y el aumento de la presión intracraneal (PIC) dan lugar al aumento de la actividad de las beta y gamma secretasas quienes transforman al β-APP en placas de Beta amiloide. Esta actividad parece comenzar unas cuatro horas tras el traumatismo y está relacionada con la aparición de la enfermedad de Alzheimer tras el TCE (10). así mismo, la inflamación crónica que se observa tras el TCE (ya sea único o repetitivo) parece favorecer este proceso, si bien es cierto que en muchos casos se observa la acumulación de esta proteína en sujetos que no desarrollaron la enfermedad, lo que sugiere que es necesaria cierta susceptibilidad individual, tal vez de carácter genético (21,22). En este sentido cabría la posibilidad de que posteriores investigaciones demostrasen que el estudio evolutivo del β-APP en su transformación en placa de -Amiloide, resultara útil también en el estudio forense del TCE, poniendo en relación muertes diferidas con traumatismos craneoencefálicos únicos o repetitivos antiguos.
El presente trabajo establece la utilidad de este marcador en el trabajo forense diario, ayudando a resolver cuestiones de trascendencia médico legal, en relación con el diagnóstico y la data de los traumatismos cráneo-encefálicos.
Materiales y métodos
Se han estudiado una serie de 25 autopsias médico legales, en 21 de las cuales la causa de la muerte fue un TCE y 4 casos de muerte súbita cardiaca consideradas como grupo control negativo. Para esto último, se eligieron muertes súbitas cardíacas evitando así que los fenómenos agónicos no provocaran fenómenos de hipoxia encefálica que artefactaran el estudio. Así mismo, no se incluyeron en el estudio casos con antecedentes de ictus, convulsiones, epilepsia, hipo o hiperglucemia.
La muestra estuvo compuesta de 11 hombres y 10 mujeres con edades comprendidas entre los 33 y los 94 años. El tiempo de supervivencia transcurrido entre el momento del TCE y la muerte oscilaba entre los pocos minutos y los 218 días.
Los casos fueron agrupados en función del tiempo de supervivencia en tres grupos: Grupo 1: menos de 2 horas de supervivencia, grupo 2: entre dos y 24 horas y Grupo 3: más de 24 horas
Las muestras fueron extraídas dentro de las 24 horas siguientes al fallecimiento. Durante este tiempo, los cadáveres se conservaron refrigerados a 4º.C.
En todos los casos se realizó autopsia completa prestando especial atención a la existencia o no de lesiones macroscópicas compatibles con la existencia de TCE (fracturas craneales, hemorragias intracraneales y procesos iatrogénicos) y se estudió exhaustivamente la documentación clínica en los casos en los que existió asistencia médica.
Una vez el cerebro ha sido extraído y pesado, se practicaron cortes de entre 1 y 1,5 cm de ancho y se obtuvieron muestras de las regiones contundidas. Estas muestras se fijaron en formol al 10% durante un tiempo nunca superior a 24 h., tras lo cual fueron incluidas den parafina con la rutina habitual. Se hicieron secciones de 4μ que se depositaron en portas silanizados. (Dako, Glostrup, Denmark).
Previamente al procedimiento inmunohistoquímico, se realizaron técnicas rutinarias con Hematoxilina-Eosina: desparafinado e hidratación Las secciones se pasan sucesivamente por: xilol, etanol 100° etanol 96°, etanol 70°, agua corriente y finalmente hematoxilina durante 5 min y tras lavar con agua corriente se pasaron por eosina 3 min
Técnica inmunohistoquímica
Primero se procedió a la recuperación antigénica calentando los potas en el microondas durante 20 min. a 750 W en tampón Tris-EDTA (Tris 10mM, EDTA 1mM, pH9). Después de lavar los portas con PBS (Buffered Saline Phosphate, 100 mM de fosfato pH 7,2; 150 mM de NaCl), las muestras fueron incubadas consecutivamente en: 1)anticuerpo primario (1h.) β-APP (Invitrogen, CA, USA, 1:50); 2) bloqueante de la peroxidasa (Dako) 10 min. 3) EnVision doble conejo, ratón (Dako) 30 min., 4) diaminobenzidina. Líquido DAB+sistema de sustrato cromógeno (Dako) 10 min. tal y como recomiendan las instrucciones comerciales (50 μL DAB en 1mL de tampón). Después de cada paso, las secciones se lavaron dos veces durante 5 min. con PBS, y tras el DAB, se lavaron con agua destilada.
La solución de anticuerpo primario β-APP se preparó con un diluyente comercial de anticuerpo (Dako). En cada caso, se incluyó un control negativo, en el cual el anticuerpo primario fue sustituido por PBS. Al final las secciones fueron contrateñidas con Hematoxilina de Harris durante 30 segundos.
Resultados
Los hallazgos macroscópicos variaron desde el edema y el piqueteado hemorrágico en córtex (tanto por contusión directa como por contragolpe) en los casos de TCE leve hasta la presencia de hemorragias intracraneales de cualquier tipo, propias del TCE de alta intensidad
En todos aquellos en los que el TCE fue de alta intensidad, el estudio del parénquima con hematoxilina eosina mostró cambios como eosinofilia, signo de desmielinización y necrosis; presencia de neuronas con un núcleo desorganizado, gránulos eosinofílicos en su citoplasma y un anillo de retracción a su alrededor (neuronas fantasma); neuronas que mostraban cambios nucleares sugerentes de apoptosis y focos de gliosis.
La cantidad y extensión de estos fenómenos, dependió del tiempo transcurrido entre el traumatismo y la muerte. En cualquier caso, los hallazgos encontrados con las técnicas convencionales no se clasificaron, pues sólo se realizaron como estudio preliminar a la selección de casos para su estudio inmunohistoquímico.
Inmunohistoquímica
Grupo control: (n=4): Dado que se tomaron como casos control pacientes que no sufrían daño neurológico de ningún tipo, la tinción resultó negativa en todos ellos Grupo problema (n= 21): En todos los casos el marcador resultó positivo en mayor o menor medida. La intensidad de la tinción varió desde pequeños puntos pálidos dispersos por el córtex a axones arrosariados y bulbos de retracción en sustancia blanca. Las variaciones en la intensidad de la tinción estaban en clara relación con el tiempo de supervivencia tras el traumatismo (Tabla 1)
Tabla 1 Evolución de la expresión del β-APP según el tiempo de supervivencia.
| Menos de 2h | 2-24 h | Más de 24 h. | ||
| Somas dispersos | SB* | |||
| SB** | + | ++ | ++ | |
| Axones arrosariados | SB | + | ++ | |
| SG | ++ | |||
| Buldos de retracción | SB | -/+ | ++ | |
| SG | ++ | |||
| Patrón de expresión | SB | Difuso | Zig-zag | |
| SG | Difuso | Difuso |
* Sustancia Blanca
**Sustancia Gris
La expresión del β-APP no presenta siempre el mismo patrón morfológico. Pueden observarse pequeños ovillos o parches dispersos por el tejido o pueden observarse axones dañados más o menos edematosos o arrosariados pero que dibujan el tejido unas zonas lesionadas en diagonal seguidas de otras franjas indemnes, dando lugar a un patrón en zigzag o ''Z''. También es posible observar un mezcla de ambos patrones.
En nuestro estudio, el patrón morfológico más común fue el difuso, pero también hubo casos en los que pudo observarse un patrón mixto: algunas regiones mostraban un patrón en zigzag, y otras mostraban un patrón difuso multifocal. Estos diferentes patrones tienen correlación con el origen primario (directamente provocado por el trauma) o secundario (al aumento de la presión intracraneal y a la hipoxia) de la lesión que estemos observando. (Fig 1)
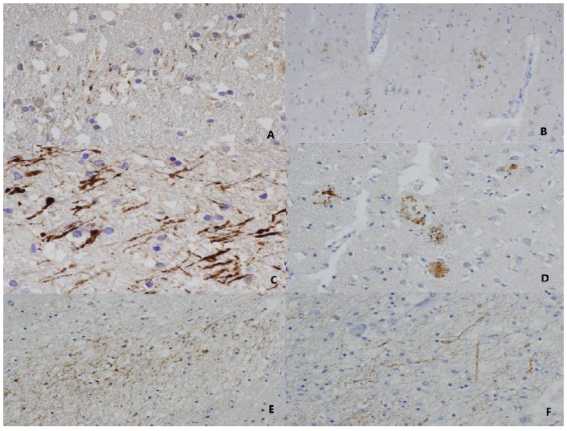
Figura 1 A: Patrón en zigzag en un caso de 30-60 min. de supervivencia. Sustancia gris.40X B: Patrón multifocal en un caso de 2h. de supervivencia. Sustancia gris.10X C: Patrón en zigzag axones arrosariados y edematosos. Bulbos axonales distales. Caso de 5 días de supervivencia. Sustancia blanca. 40X. D: Patrón multifocal en un caso de 26 días de supervivencia. Sustancia gris 20X. E: Región multifocal de un patrón mixto en un caso de 17 días de supervivencia. Sustancia blanca 20X F: Zona en zigzag de un patrón mixto, en ese mismo caso de 17 días de supervivencia. Sustancia blanca. 20X. Se observa el aumento de intensidad y los cambios progresivos conforme aumenta el tiempo de supervivencia. En el caso de 17 días se distingue el daño primario del secundario por los distintos patrones de distribución en distintas áreas del córtex
En casos de muertes diferidas por TCE, es decir, aquellos que sobrevivieron durante semanas o meses en el hospital, se pudo incluso comprobar como este marcador llega a negativizarse, conforme avanza la astrogliosis.
Discusión
Nuestros primeros resultados coinciden con lo observado por distintos autores (14-18) ya que sólo se observa positividad a este marcador cuando existe un cuadro inflamatorio cerebral, siendo completamente negativo en los casos control en los que no existió afectación neurológica en el mecanismo fisiopatológico de la muerte.
Así mismo en los casos con una supervivencia de varios meses, encontramos que este marcador también se vuelve negativo, conforme avanza la cicatriz glial en consonancia con lo observado por Onyszchuk (23) Oehmichem (24) y Willemsee-van Son(25) . Por eso, los casos de muerte diferida por TCE grave ocurrida en el hospital tras varias semanas o incluso meses fueron descartados ya que, además, carecen de interés médico forense al quedar documentadas detalladamente en la historia clínica por lo que establecer las circunstancias y causas de la muerte no presenta dudas.
Patrones de distribución: utilidad del b-app como herramienta diagnóstica (hipoxia versus trauma)
En primer lugar, cabe señalar que la patología amiloidea es distinta en distintas poblaciones de TCE, porque el daño cerebral es espacialmente heterogéneo, dependiendo del tipo de trauma, su severidad y la localización del impacto.
Una clasificación sencilla de la patología resultante del TCE es aquella que distingue el daño cerebral en focal y difuso:
El daño focal incluye contusiones que afectan la superficie del cerebro, es decir, aquellos TCE que cursan con hemorragias intracraneales y/o aumento de la presión intracraneal.
El daño axonal difuso (DAI) se caracteriza por disrupciones en los axones que componen la sustancia blanca, y se debe a fuerzas rotacionales y de aceleración y desaceleración. (26)
Todo esto sugiere que el tipo de insulto también va a tener un papel importante en la dinámica de la acumulación de -APP. (27)
De hecho, en modelos experimentales que se centraban en el DAI y que emplean fuerzas rotacionales a distintas revoluciones por segundo, se observó distinta expresión focal y cronológica de las proteínas amiloideas en cada grupo (28). Así mismo, también existen diferencias entre autores que emplean mecanismos de contusión por objeto contundente y los que emplean un fluido a presión para provocar el insulto cerebral (22)
Por otro lado, la hipoxia per se no es causa de daño axonal, sino que éste se produce de forma secundaria a complicaciones vasculares causadas por el aumento de la presión intracraneal y la herniación del cerebro. Dado que el transporte de β-APP es activo (proceso dependiente de energía), es razonable asumir que también se acumula en casos de compromiso metabólico grave tales como hipoglucemia, hipoxia global, infección y/o edema cerebral (29).
El estudio del β-APP permite también diagnosticar la existencia de aumento de la presión intracraneal (incluso cuando sus niveles no han sido tan elevados como para causar herniaciones). El origen de este problema (bien por hipoxia de origen cardíaco, intoxicación por CO o epilepsia, bien por una alteración metabólica), habitualmente puede ser determinado por los antecedentes clínicos del sujeto (30).
Dado que la acumulación de esta proteína no se debe a un solo mecanismo lesional, algunos autores (3,29) han intentado encontrar características que permitan diferenciar las distintas causas. El patrón de distribución de β-APP puede resultar útil para evitar falsos positivos a la hora de etiquetar como traumática una muerte. Así podemos encontrar tres grandes patrones de distribución:
Patrón difuso multifocal: presentan edemas axonales en la región traumatizada, y a distancia por lesiones de contragolpe y por el edema cerebral.
Patrón en zigzag o Z: se produce en regiones perivasculares limítrofes a una región de isquemia o infarto.
Patrón mixto: Son grupos de axones con trauma difuso y otras zonas en Z por la hipoxia secundaria al aumento de la presión intracraneal. Estos patrones se encuentran sobre todo en las estructuras de la línea media encefálica, especialmente en el cuerpo calloso y en el puente, aunque pueden observarse también en el córtex.
Así en general, se admite que el patrón difuso multifocal de varicosidades axonales se asocia con un origen traumático del daño axonal, el patrón en zigzag con daño por hipoxia y el patrón mixto se relaciona con traumatismos severos en los que el edema cerebral o alteraciones respiratorias añaden un componente hipóxico al traumático.
Esto concuerda con el presente estudio, en el que se observa el cambio de patrón de expresión (en especial en la sustancia gris) conforme aumenta el tiempo de supervivencia, ya que es necesario un tiempo mínimo para que se establezcan esos fenómenos inflamatorios y de edema.
Onyszchuk (23) establece en casos de corta supervivencia (no superior a 2 horas) se describen distintos grados de tinción en relación con la severidad del traumatismo que van desde la negatividad para este marcador, hasta la aparición masiva de bulbos axonales
En los grados 0-2, la expresión inmunohistoquímica puede ser muy débil y su origen puede ser isquémico. El tiempo de supervivencia determina la evolución del daño axonal y el grado de tinción, siendo casi imposible detectar más de un grado 2 en casos con una supervivencia inferior a 35 minutos.
Nuestro trabajo no es útil para de diagnosticar la extensión del daño secundario, porque a diferencia otras investigaciones, no se trata de condiciones experimentales, sino que pretende aproximarse a la práctica médico forense habitual, por lo que sólo estudiamos la región del impacto, lo que limita el diagnóstico de la extensión de la hipoxia, que es lo que probablemente detecte el estudio de Onyszchuk (23), y que puede tener importancia en la práctica clínica pero que no es tan trascendente en el diagnóstico postmortem. Por ello, dado que los cambios más precoces en este sentido se producen en la sustancia gris, el estudio de esta zona sigue siendo útil, pudiendo ser determinante para establecer la relación causa efecto de la muerte con traumatismos moderados o leves, en los que no se produce una lesión macroscópica evidente. Es decir, su presencia, puesta en un contexto clínico y forense nos permite resolver dudas diagnósticas tal como afirman también otros autores (31,32).
De hecho, existió un caso en el que este marcador fue importante para establecer el nexo de causalidad con el traumatismo causante de la muerte. Dicho caso se clasificó en el grupo de supervivencia superior a 24 horas (cinco días), pues la paciente sufrió una caída con pérdida de conciencia por la que se negó a acudir a los servicios de urgencias. Cinco días más tarde sufre otra caída en la calle, y esta vez sí que es trasladada al hospital, pero se encontraba consciente y orientada con una escala de Glasgow de 15, pero en el servicio de radiología comenzó a deteriorarse llegando a descender este índice a 3 y fallecer al poco tiempo. El TC demostró una fractura del hueso occipital derecho y una hemorragia intracraneal fatal. Dado que clínicamente fue más grave el primer traumatismo, en el que existió pérdida de conciencia, se achacó a éste el hematoma subdural, clasificándolo como subagudo. Al observar la tinción con β-APP su comportamiento se correspondía con una evolución no superior a 2h. Sin que se observara afectación axonal que sugiriera un aumento progresivo de la PIC e hipoxia secundaria, por lo que la hemorragia intracraneal era reciente y así el éxitus se relacionaba con el segundo traumatismo. Este caso no revestía trascendencia legal, pero este tipo de valoraciones puede ser crucial en casos de homicidios o accidentes.
Por otro lado, se ha propugnado que es necesario un amplio muestreo para el diagnóstico del DAI y observar simultáneamente otros cambios morfológicos que sugieran un origen isquémico, como la lesión de la zona C1 del hipocampo (que es la región del encéfalo más vulnerable a la hipoxia), la eosinofilia y necrosis neuronal (33). Otros autores proponen empelar, al menos, 6 bloques para llegar al diagnóstico (y deben abarcar los dos hemisferios) (34).
Sin embargo, estudiar muestras del área contundida nos permite determinar la severidad del traumatismo y aproximarnos al tiempo de supervivencia y tiene la ventaja de no necesitar un entrenamiento o equipamiento especiales, lo que nos permite tomarlas incluso en condiciones poco propicias. Además, la valoración del área contundida tiene un gran valor diagnóstico desde el punto de vista forense, porque los cambios en esta región son más evidentes y significativos, ya que esta es la ''zona cero'' desde la que se extiende todo el daño al resto del encéfalo (35).
Cronología de la expresión del β-app tras un traumatismo
Hay que tener en cuenta que el daño axonal y, por ende, la acumulación de proteínas amiloideas se produce en dos fases: en la fase aguda, el agente traumático provoca la ruptura axonal (en especial en el córtex) y esto es el estímulo inicial que provoca la acumulación de esta proteína. En la fase crónica del daño cerebral postraumático se produce una alteración vascular que incluye la disminución del aclaramiento de dichas proteínas, cerrando el círculo del daño axonal y provocando su extensión a regiones alejadas del foco traumático. (27)
Existe una correlación entre la positividad para β-APP y el tiempo transcurrido entre el traumatismo y la muerte. No se detecta inmunorreactividad significativa de esta proteína por debajo de los 30 minutos ya se produzca la muerte por un traumatismo o por otras causas. Además, el intervalo de tiempo en el que podemos detectar la inmunohistoquímica disminuye al aumentar la severidad del traumatismo (29).
De forma generalizada se acepta que en los casos de daño traumático (a partir de un año), el daño axonal difuso no se puede detectar antes de las 1,5 horas post-traumatismo (17).
En anteriores estudios forenses, la presencia de bulbos axonales no se observa en supervivencias menores de 6 h, momento en el que también se produce el acúmulo de β-APP y NF (neurofilamentos) en el soma (36). Lejos de la zona traumatizada, a partir del 3-7 días post-trauma se observan axones varicosos y edematosos (mediante anticuerpos anti-APP y NF) disminuyendo su número a partir de los 6 meses. Sin embargo, β-APP se ha demostrado a las 3 horas tras la lesión, y a los 6 meses permanece positivo en el parénquima y junto a los vasos (37). Otros autores, lo detectan a las primeras 24 h postraumatismo en neuronas edematosas y con distrofia de sus fibras (15).
En casos de traumatismos leves se observa un comportamiento temporal del β-APP característico: es positivo entre 1,5 horas y 3 horas tras la lesión, pero después el número de axones que expresan esta proteína aumenta hasta alcanzar su pico máximo a las 10-15 horas, estabilizándose tras este momento (14).
Las diferencias cronológicas que se observan en los distintos trabajos se deben a que la mayoría estudia la expresión de este marcador en regiones alejadas de la zona traumatizada, es decir, observan el daño secundario a fenómenos hipóxicos, y esta circunstancia puede tardar más o menos en ocurrir dependiendo de la severidad de traumatismo. Por otro lado, cada estructura encefálica muestra una sensibilidad diferente a la hipoxia por lo que también se producirán diferencias cronológicas en la expresión de este marcador en las distintas partes del encéfalo.
Así Kelley (18) estudió cómo se altera el trasporte axonal, observando el comportamiento del β-APP en el núcleo talámico (que es la estructura más sensible a la hipoxia y por tanto, al daño axonal secundario) tras producir una lesión por axotomía. A los 15 minutos observan inmunoreactividad para esta proteína en el núcleo talámico y conforme transcurre el tiempo el diámetro de los axones va aumentando y a partir de los 60 min comienzan la aparición de los bulbos axonales.
Li observó que tras un traumatismo moderado las primeras regiones donde se detecta son el tracto parasagital y las estructuras de la línea media encefálica, incluyendo la parte caudal del cuerpo calloso (38). Así, la distribución más frecuente de la inmunoreactividad para β-APP es en el tallo cerebral (95% de los casos), la cápsula interna y el tálamo (80% de los casos) el cuerpo calloso y la sustancia blanca parasagital (70% de los casos). La expresión de β-APP es mayor que la presencia de hemorragias, por lo que se ha sugerido que la expresión del β-APP no depende de la existencia de hemorragia para su acumulación. (14). Esto es consistente con nuestro trabajo, ya que, independientemente de la severidad del traumatismo observábamos la expresión de este marcador en regiones que presentaban signos superficiales o externos de contusión.
En la protuberancia, la positividad para β-APP no es evidenciable antes de 3h de supervivencia (lo cual demostraría la vitalidad de la lesión), independientemente de si ha existido o no hematoma subdural y/o hemorragia cortical, incluso se produce en casos de trauma no mecánico. En casos de hipoxia/isquemia la positividad es más tardía (8-24h) y su expresión es mucho menor que en causas traumáticas.
El daño axonal puede demostrarse en la protuberancia empleando como marcador el β-APP sin diferencias significativas según la etiología. Muchas veces, simplemente es una consecuencia tardía de cambios complejos del citoesqueleto debido a un episodio traumático, lo que conduce a un colapso del transporte axoplasmático anterógrado. Evoluciona a edema axonal, varicosidades y bulbos y por último a la desconexión tal y como ya se ha expuesto. No obstante, la distribución topográfica del daño axonal a lo largo de las estructuras de la línea media del cerebro probablemente nos oriente en lo que al diagnóstico del tipo de mecanismo lesional se refiere (24).
Por otro lado, la gravedad de la expresión histopatológica traducida en el diámetro de los axones edematosos depende de diversos factores como el tiempo transcurrido desde el impacto, el tamaño original del axón, el tiempo de acción de los efectos secundarios y el tiempo que puede tomar cualquier mecanismo potencial de regeneración (25)
Por último, el daño axonal puede comenzar incluso 84 horas después del traumatismo, lo que refleja un daño continuo secundario. Además, la inmunoreactividad positiva continúa meses tras el agente causal (pudiendo llegar a provocar enfermedades neurodegenerativas), lo que indicaría un mantenimiento persistente del flujo axonal rápido. También es observable un periodo de regeneración significativa, de lo que se deduce que, hasta cierto punto y en casos leves, el daño axonal leve puede ser reversible (39).
Otro factor que influye en la rapidez con la que se expresa el β-APP es el diámetro de los axones. Se observó que en el quiasma óptico y en el cuerpo calloso, el pico máximo de expresión se producía a las 8 h en el quiasma y a las 28 h en el cuerpo calloso, cuyos axones son de menor calibre y más ricos en microtúbulos, mientras que los primeros tienen mayor diámetro y poseen mayor densidad de neurofilamentos. (40)
En resumen y de forma generalizada el proceso (activo) de formación de beta APP se observa de forma evidente a las 24 horas, presenta una meseta de actividad a los 10-14 días, a partir de los cuales comienza a disminuir. Mediante inmunohistoquímica los primeros cambios en el tránsito del β-APP (en axones edematosos) se demostraron a los 30 minutos y duran hasta semanas. Se puede considerar positivo a la primera hora, y no solo en la zona contundida, sino también en zonas lejanas al punto de impacto. El pico máximo se produce a las 48 horas y continúa hasta dos semanas en la sustancia blanca bilateral, hipocampo y córtex. En el tallo cerebral permanece durante una semana (41,42).
Una vez más, el hecho de reproducir condiciones reales de trabajo, en las que se desconoce la intensidad del impacto y en las que no resulta práctico un muestreo tan amplio, limita la interpretación de nuestros resultados. De la misma manera que los problemas con la sensibilidad de este anticuerpo, como le ocurrió a otros investigadores (24,43,44). No obstante, nuestros resultados no difieren del consenso general, en especial en lo que se refiere al periodo penumbra de las dos primeras horas tras el traumatismo que es el que más problemas suele plantear desde el punto de vista médico legal, en especial en traumatismos moderados o leves, observando somas neuronales teñidos aunque sea de forma débil en traumatismos de entre 30 min y dos horas. Por otro lado, en los casos de muertes diferidas tras un traumatismo leve o moderado bien único o bien repetitivo, el patrón difuso o en zigzag nos permite establecer la relación causa efecto entre el traumatismo y la muerte,aunque hayan transcurrido 24 horas o más desde el evento traumático.
Conclusión
El β-APP es un marcador ampliamente estudiado tanto con propósitos clínicos como pronósticos, sobre todo en relación con las secuelas a largo plazo del TBI,. En nuestro estudio ponemos de manifiesto que también es útil en la práctica forense. En primer lugar, porque es una técnica asequible, segundo, porque al ser una proteína que no se expresa en condiciones fisiológicas, su simple presencia ya significa que existe daño neuronal.
Nuestro trabajo se basa en las condiciones habituales del trabajo del médico forense, por lo que se simplifica la toma de muestras y su interpretación. Así el tipo de alteraciones que se observen con este marcador (tinción de somas neuronales, axones arrosariados y bulbos axonales) nos permite acotar el tiempo de evolución, y el patrón de expresión (difuso o en zigzag) nos permite distinguir el daño directo provocado por el trauma del daño secundario provocado por la hipoxia.
Dado que todos estos fenómenos son independientes de la naturaleza e intensidad del traumatismo (aunque se expresen con menor intensidad en los casos en los que no existió hemorragia intracraneal), este marcador resulta de gran utilidad para establecer nexo de causalidad entre traumatismos moderados y leves, con la muerte diferida del sujeto, ayudando a resolver casos de trascendencia médico legal.

