











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción.
El síndrome metabólico es un desorden complejo con un alto costo socioeconómico y se le considera un problema de salud pública, pues constituye una verdadera epidemia 1.
En general la IDF (Federación Internacional de Diabetes) estima que un cuarto de la población mundial tiene SM, aunque la prevalencia va desde menos del 10% hasta un 84%, dependiendo de la región, urbana o rural, composición (sexo, edad y etnia) de la población estudiada y según la definición de SM utilizada 2.
La presencia del SM en un individuo aumenta el riesgo de padecer de diabetes mellitus tipo 2 y/o de enfermedad cardiovascular.
El propósito de este trabajo es revisar el tema del síndrome metabólico desarrollando varios aspectos del mismo: definiciones, epidemiología, etiología, componentes y tratamiento.
Definición.
El síndrome metabólico (SM) se considera un estado fisiopatológico crónico y progresivo, que representa a un grupo de factores de riesgo (obesidad, resistencia a la insulina, hipertensión y dislipidemia principalmente) que forman un síndrome complejo definido por una patofisiología unificadora y que se asocia con un riesgo aumentado para la enfermedad cardiovascular (ECV), diabetes mellitus tipo 2 y otros desórdenes relacionados 3-5.
Según Kaur el SM en pacientes aumenta en cinco veces el riesgo de sufrir diabetes mellitus tipo 2 y en dos veces el riesgo de desarrollar una ECV en los próximos 5 a 10 años comparados con individuos sin SM. Además, los pacientes con dicho síndrome tienen un riesgo de dos a cuatro veces de sufrir derrame cerebral y de tres a cuatro veces de sufrir infarto al miocardio 2.
La etiología de la ECV en pacientes con SM puede involucrar: enfermedad aterosclerótica coronaria, hipertensión arterial, hipertrofia del ventrículo izquierdo, disfunción diastólica, disfunción endotelial, enfermedad micro-vascular coronaria y disfunción autonómica. La patogénesis de la ECV en el SM es multifactorial y puede ser causada por uno o más factores asociados con esta condición tales como la resistencia a la insulina, la diabetes o la inflamación crónica. Una característica común de la ECV en el SM y la resistencia a la insulina es la presencia de estrés oxidativo aumentado en el corazón 6.
Hutcheson y Rocic dan cuenta que la presencia de obesidad abdominal con 2 o más componentes del SM sin hiperglicemia resulta en una incidencia elevada (2.5 veces) de engrosamiento de la media-intima de la carótida, un indicador temprano de aterosclerosis subclínica, mientras en aquellos con hiperglicemia la incidencia es de 6 veces. La glucosa aumentada en un trasfondo de obesidad abdominal correlaciona fuertemente con el desarrollo de la enfermedad arterial coronaria en mujeres, mientras que un HDL-colesterol al lado de la obesidad abdominal es un predictor excelente de la enfermedad arterial coronaria. Aún la obesidad abdominal, en ausencia de cualquier otro componente del SM, parece predecir el riesgo cardiovascular futuro en hombres 7. Estos dos investigadores indican, además, que la etiología de la ECV parece estar relacionada con el estrés oxidativo del SM.
El SM incrementa la probabilidad de ECV a un mayor grado que la probabilidad conferida por cualquiera de sus componentes individuales 7. Todos los componentes están causalmente inter-relacionados y cada componente contribuye independientemente a un riesgo aumentado de ECV 8.
Definiciones internacionales.
Desde que el SM fue descrito por Raven en 1988, varias definiciones han sido publicadas y revisadas y numerosos estudios han explorado su patofisiología. El primer intento fue hecho en 1998 por parte de la OMS (Organización Mundial de la Salud) y poco después le siguió la EGIR (Grupo europeo para el estudio de la resistencia a la insulina) y posteriormente han surgido otras definiciones de otras organizaciones internacionales 1.
La definición más ampliamente utilizado por su sencillez es la de la NCEP-ATP III (del inglés, “National Cholesterol Education Program Adult Treatment Panel III”) (Tabla1). Bajo esta definición todos los parámetros tienen el mismo valor, no se enfatiza en ningún factor de riesgo o componente. Bajo las directrices de la Asociación Americana de Diabetes (ADA, del inglés “American Diabetes Association”) la definición del factor disglicémico fue cambiada para la glucosa en ayunas (de ≥ 110 mg/dl a ≥ 100 mg/dl) en la definición de SM por la NCEP-ATP III 9.
La definición de la AHA/NHLBI es muy similar a la de la NCEP-ATP III, todos los factores de riesgo tienen la misma importancia y los valores son los mismos (Tabla 1).
En 3 de las clasificaciones (OMS, EGIR y AACE) se enfatiza en la presencia de la insulino resistencia (IR). En las tres definiciones mencionadas tanto la TGA (Tolerancia a la glucosa alterada) como la GAA (Glucosa alterada en ayunas) son marcadores de un metabolismo anormal de la glucosa. Ninguna de las tres definiciones incluye a la diabetes mellitus (Tabla 1).
En estas tres definiciones citadas anteriormente se enfatiza el papel de la IR, pues en su ausencia no hay SM, aunque el individuo tenga o cumpla todos los demás criterios.
Para la estimación de la sensibilidad o la resistencia a la insulina el patrón de oro es el “clamp euglicémicohiperinsulinémico.
Sin embargo es una técnica compleja que ha hecho que se prefiera el uso del HOMA (del inglés “Homeostasis Model Assessment”). Esta última técnica ha sido probada y constituye una alternativa válida como método de diagnóstico de la IR 10. Frankenberg y colaboradores utilizaron el método HOMA para estudiar la IR 11.
El método HOMA deriva de un estimado de la sensibilidad de la insulina partir de un modelo matemático de las concentraciones plasmáticas de glucosa e insulina en ayunas. Los resultados altos de HOMA denotan resistencia a la insulina alta 12.
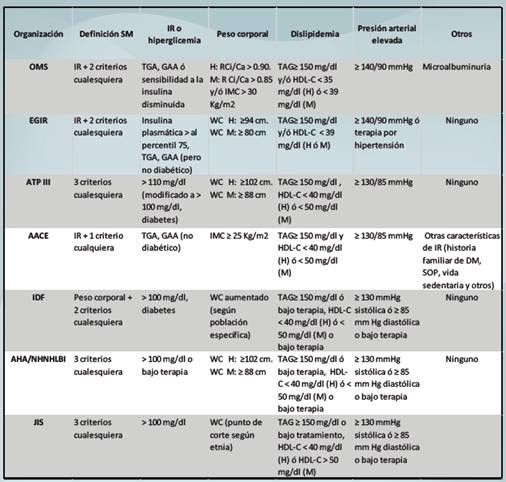
AACE: Asociación Americana de Endocrinólogos; AHA: Asociación Americana del Corazón; ATP III: Panel III del Tratamiento de Adultos del Programa Nacional de Educación del Colesterol; Ca: cadera; Ci: cintura; EGIR: Grupo Europeo para el Estudio de la Resistencia a la Insulina; GAA: Glucosa alterada en ayunas; H: hombres; HDL-C: colesterol HDL; IDF: Federación Internacional de Diabetes; IMC: Índice de masa corporal; IR: insulino resistencia; JIS: Declaración Provisional Conjunta; M: mujeres; NHLBI: Instituto Nacional de la Sangre, del Corazón y del Pulmón; OMS: Organización Mundial de la Salud; R: relación; SOP: síndrome de ovario poliquístico; TAG: Triglicéridos; TGA: Tolerancia de la glucosa alterada; WC: circunferencia de la cintura.
En el caso de la IDF el énfasis es en la obesidad, determinada por la circunferencia de la cintura y cuyo valor es específico de la población y etnia estudiadas 13.
La última definición, JIS (del inglés “Joint Interim Statement”), fue propuesta en 2009 por varias organizaciones tales como la AHA/NHLBI, IDF, la Sociedad Internacional de Aterosclerosis y otras. Estas organizaciones intentaron armonizar los criterios de los diferentes componentes del SM y dar una definición unificada: una serie individual de puntos de corte únicos para todos los componentes excepto para la circunferencia de la cintura (pueden utilizarse puntos de corte nacionales o regionales) 14-16. No obstante, el grupo de trabajo decidió explorar a futuro la relación de los umbrales de la circunferencia de la cintura y los riesgos cardiovasculares en diferentes poblaciones.
Es de destacar que para estimar la obesidad se recurre a diferentes determinaciones: relación cintura / cadera (OMS), circunferencia de la cintura (EGIR, NCEP-ATP III, IDF y AHA/NHLBI) o IMC (AACE). El punto de corte para la circunferencia de la cintura es variable y depende de la etnia y del sexo.
Así por ejemplo Al-Aqeedi y colaboradores en un estudio sobre la prevalencia del SM en pacientes masculinos hospitalizados por síndrome coronario agudo, utilizaron los puntos de corte de circunferencia de la cintura para la población asiática (≥ 90 cm) y medio-este y mediterránea masculina (≥ 94 cm) 17.
En otro estudio de prevalencia del SM en Irán se utilizó un punto de corte de ≥ 95 cm para ambos sexos 18.
Sperling y colaboradores indican que la medición de la circunferencia de la cintura incluye tanto los depósitos de grasa visceral como subcutánea abdominal y estos dos depósitos son anatómica y fisiológicamente diferentes, especialmente dentro de la población obesa. La grasa visceral está asociada con prediabetes y diabetes tipo 2, hipertensión y a un mayor riesgo de ECV 5. No obstante, estos mismos investigadores hacen la aclaración que la circunferencia de la cintura es un mejor predictor de la grasa corporal total que el IMC.
Los puntos de corte para la circunferencia de la cintura establecidos tampoco son aceptados universalmente. Ogawa y colaboradores, basándose en estudios realizados en diferentes países asiáticos, indican que los puntos de corte de la NCEP-ATP III deberían ser menores para las poblaciones asiáticas. Además, que los puntos de corte establecidos por la IDF para las poblaciones asiáticas (90 cm para hombres y 80 cm para mujeres) no son los óptimos basados en varios estudios realizados en India, China, Singapur y Corea 19.
Bener y colaboradores compararon cuatro medidas utilizadas para determinar el índice de obesidad, IMC, circunferencia de la cintura, relación cintura/cadera y la relación cintura/altura en la población adulta qatarí. Los investigadores llegaron a la conclusión que la circunferencia de la cintura a un punto de corte de 99.5 cm en hombres y 91 cm en mujeres era el mejor predictor de SM 20.
Otra variación en la estimación de la obesidad es el índice de la obesidad central (ICO, del inglés “index of central obesity”). Este índice es la relación de la circunferencia de la cintura (WC) con la altura y muestra una buena correlación con la adiposidad central y es un buen predictor de diabetes tipo 2. Además, tiene una fuerte correlación con los niveles de la hormona leptina, con el perfil lipídico aterogénico, con el estrés oxidativo y con el riesgo cardiovascular.
También es útil para detectar obesidad central en la infancia y la resistencia a la insulina en los niños 21. Según dos investigadores indios el ICO podría reemplazar a la WC en todas las definiciones del SM y podría obviarse de este modo las variaciones por etnicidad y género en los puntos de corte.
Así mismo Zhou y colaboradores evaluaron la idoneidad de la circunferencia del cuello como índice de obesidad y predictor del SM o de riesgo cardiovascular en la población china. Los investigadores compararon esta medida contra otras medidas o índices tradicionales, IMC, circunferencia de la cintura, la relación cintura/cadera y la relación cadera/altura. En sus conclusiones establecen que la medida de la circunferencia del cuello es simple e identifica el SM y puede ser utilizada como una herramienta de tamizaje para el SM y otras enfermedades crónicas asociadas a la obesidad 22.
En dos definiciones se incluye a la diabetes mellitus dentro de los factores de riesgo (NCEP-ATP III e IDF). No obstante, la inclusión de la diabetes tipo 2 es materia de debate. Para Shin y colaboradores la inclusión de la diabetes dentro de los parámetros del síndrome, hace que el mismo pierda su ventaja clínica como un predictor para el desarrollo de la diabetes 9.
Prevalencia del sm según la definición.
Ante el hecho de la existencia de varias definiciones del SM la prevalencia del mismo varía con la definición utilizada, aunque lo que sí se observa es una mayor prevalencia del SM en las mujeres independientemente de la definición utilizada 18,23,24.
Wen y colaboradores compararon la prevalencia del SM en población china rural utilizando las definiciones de la OMS, IDF, NCEP-ATP III y JIS. La prevalencia en hombres era 11.5% (OMS), 32.4% (NCEP-ATP III), 27.5% (IDF) y 47.7%
(JIS). En las mujeres era de 15.7% (OMS), 54.2% (NCEP-ATP III), 51.5% (IDF) y 54.2 (JIS) 15. Se nota una mayor diferencia en los valores de los hombres.
En otro estudio, realizado en regiones urbanas y rurales de Irán, Khosravi-Boroujeni y colaboradores compararon las prevalencias obtenidas utilizando diferentes definiciones del SM. Las prevalencias en hombres fueron 10.6 (OMS), 20.7% (NCEP-ATP III), 25.5% (IDF), 29.1% (JIS), 6.9% (EGIR) y 21.4% (AHA-NHBI). En las mujeres fueron de 15.9%
(OMS), 38.8% (NCEP-ATP III), 30.2% (IDF), 33.2% (JIS), 10.7% (EGIR) y 51.7% (AHA-NHBI) 18.
En otro estudio, efectuado en Sri Lanka, se compararon las prevalencias del SM utilizando las definiciones de NCEPATP III e IDF. Las prevalencias obtenidas fueron: en hombres 22.9% (IDF) y 33.9% (NCEP-ATP III) y en mujeres45.8% (IDF) y 56.1% NCEP-ATP III) 25.
Por último, se cita la prevalencia obtenida en población urbana lituana utilizando diferentes definiciones. La prevalencia en los hombres fue de 27.2% (NCEP-ATP III), 33.4% (AHA/NHLBI), 39.4% (IDF) y 44.1 (JIS). En las mujeres fue de 33.9%, 41.5%, 47.2 y 48.7% respectivamente 14.
Peso de los componentes y efecto aditivo.
Todos los componentes del SM de las varias definiciones existentes están involucrados en el riesgo cardiovascular y de diabetes tipo 2. En particular, los componentes aterogénicos (HDL-C bajo y TAG elevados) están asociados individualmente al riesgo cardiovascular, mientras que la IR incrementa el riesgo de desarrollar diabetes tipo 2. La obesidad central se asocia con riesgo aumentado de enfermedad cardiovascular y de diabetes tipo 2 1.
Diversos estudios han mostrado que no todos los componentes individuales del SM contribuyen igualmente al riesgo de ECV o de mortalidad por cualquier causa.
En un estudio en China el riesgo aumentado de mortalidad por cualquier causa era predicho significativamente por una glucosa en ayunas alterada (GAA) en todos los sujetos 24 y en otro estudio en Italia dicho riesgo era mejor predicho por el HDL-C bajo y la GAA en mujeres en una población italiana 26.
Khosravi-Boroujeni y colaboradores en un estudio realizado con poblaciones urbana y rural de Irán encontraron que el riesgo de enfermedad cardiovascular era mayor para la intolerancia a la glucosa o la diabetes que para cualquier otro componente 18.
Wen y colaboradores señalan que algunos componentes individuales son determinantes más importantes de enfermedad arterial periférica (EAP) y de derrame cerebral 15. Así por ejemplo el HDL-C bajo no estaba asociado a EAP en mujeres y todos los índices dislipidémicos estaban asociados con derrame cerebral en hombres, pero no en las mujeres.
Igualmente, en otro estudio realizado en Corea del Sur se concluyó que el HDL-C y la glucosa en ayunas, pero no los TAG ni la presión arterial o el IMC, eran predictivos de la enfermedad arterial coronaria luego de ajustar los análisis 27.
Finalmente, unos investigadores croatas encontraron que la presión arterial sistólica era un predictor más fuerte, entre todos los componentes del SM, para el derrame cerebral 28.
Otro aspecto a considerar es el efecto sobre el riesgo entre más factores se tengan presentes. En un estudio realizado por Wilson y colaboradores el riesgo relativo para diabetes tipo 2 se incrementaba con el número de componentes del síndrome cuando se utilizaba la definición del NCEP-ATP III 29.
En un estudio en Corea del Sur donde se analizó la participación de los componentes del SM se concluye que incluso teniendo solo 1 ó 2 factores (que se considera no hay SM) hay un riesgo aumentado de desarrollar tanto la enfermedad cardiovascular aterosclerótica como de enfermedad cardíaca isquémica. El riesgo era de 1.5 y 2.3 veces más alto para 1 ó 2 factores respectivamente 27.
En otro estudio efectuado en Irán se notó que entre más componentes del SM se sumaran mayor es el riesgo de eventos cardiovasculares. El riesgo cardiovascular para individuos teniendo 4 ó 5 factores era de 2.98 y 6.06 respectivamente 18.
Utilizando población japonesa de mediana edad y con la definición de la OMS el riesgo de incidencia de enfermedad cardiovascular fue de 3.18, 3.48, 12.55 y 14.15 para la presencia de 1,2, 3 y ≥ 4 componentes del SM 30. El riesgo relativo de incidencia de diabetes tipo 2 fue de 1.92, 4.36, 6.44 y 15.08 respectivamente.
La adiponectina es una hormona que sensibiliza a los hepatocitos, promueve la oxidación de los ácidos grasos en el hígado, disminuye el nivel de TAG e incrementa la captación de glucosa por el músculo esquelético. Frankenberg y colaboradores encontraron que los niveles de esta hormona disminuyen según se incrementa el número de componentes del SM involucrados 11. Esta sería una forma como el SM podría influir sobre el riesgo cardiovascular o el riego de diabetes tipo 2, disminuyendo el nivel de alguna hormona que tiene efecto sensibilizante a la insulina.
Epidemiología.
La presencia del SM se asocia con la edad, con la actividad física disminuida, con la dislipidemia, la hipertensión, con el tratamiento con antihiperglicémicos orales y con los niveles de HBA1c ≥ 7.0%. El riesgo es mayor en las mujeres, en los pacientes con niveles elevados de glucosa en ayunas y en los desórdenes endocrinos 13.
Varios factores modulan la prevalencia del SM en la presencia de obesidad, incluyendo una calidad nutricional pobre y la carencia de actividad física. Monteiro y Azevedo indican que la grasa saturada y la grasa dietaria total están asociadas con la IR y con la presión arterial elevada como también con la inflamación asociada a la obesidad.
El consumo de una dieta rica en ácidos grasos saturados resulta en un perfil de expresión génica proinflamatoria asociada a la obesidad, mientras que el consumo de una dieta rica en ácidos grasos monoinsaturados se asocia a un perfil más antiinflamatorio 31.
La emergencia de la obesidad y del SM en países en desarrollo se relaciona con un número de factores. La transición demográfica (hacia menor fertilidad, menor mortalidad y mayor expectativa de vida) y la transición epidemiológica (de una prevalencia por enfermedades infecciosas a un patrón de enfermedades relacionadas al estilo de vida) han llevado a cambios significativos en la dieta y en la actividad física. Estos cambios causan efectos significativos sobre la composición corporal y el metabolismo, resultando frecuentemente en aumentos en el IMC, en la obesidad abdominal, en la dislipidemia y en la diabetes tipo 2 1.
La edad, la etnicidad y el sexo contribuyen a la susceptibilidad metabólica, en parte mediada por diferencias en la distribución del tejido adiposo y del tamaño del adipocito. Por ejemplo, los asiáticos del sur tienen un contenido de grasa corporal, una relación cintura / cadera, una relación grasa visceral / grasa subcutánea y un área de los adipocitos mayor que los caucásicos ajustado por edad, sexo e IMC 32,33.
Otros factores asociados a la emergencia del SM incluyen el fumado, la historia familiar de diabetes, el nivel socioeconómico y el nivel educativo 2.
El IMC continúa siendo uno de los factores de más peso para explicar la prevalencia e incidencia del SM y precisamente la NHANES (del inglés “National Health and Nutrition Examination Survey”) indica que 5% de los sujetos con peso normal tienen SM, contra un 22% en sujetos con sobrepeso y un 60% entre las personas obesas 34. Además, los hombres y mujeres obesas tienen más de 6 veces y 5.5 veces, respectivamente, de cumplir los criterios del SM que sus contrapartes de peso normal 1.
Etiología del SM.
Subyacente al SM y a varios de sus componentes se encuentra un proceso inflamatorio crónico subclínico. Un grupo de investigadores italianos al tratar de establecer la patogénesis del SM establecen su origen en la obesidad y en el sobrepeso. Ambos estados, y especialmente la obesidad, se asocian a una inflamación crónica de bajo grado que juega un papel importante en el desarrollo de la IR. Finalmente, la IR vendría a ser el disparador de las comorbilidades asociadas al síndrome metabólico, tales como aterosclerosis, dislipidemia, hipertensión, un estado protrombótico e hiperglicemia 35. La asociación entre la obesidad y la inflamación se da por medio de la sobreproducción de citoquin proinflamatorias por parte del tejido adiposo acumulado en exceso. En este modelo la sobrealimentación es vista como una injuria celular y la respuesta de las células metabólicamente activas, como el adipocito, es una liberación de factores proinflamatorios dando inicio al proceso inflamatorio mencionado.
No obstante, aunque la obesidad y la IR están en el centro de la patofisiología del SM, un número de otros factores tales como el estrés crónico y la regulación alterada del eje hipotálamo-hipófisis-adrenal y del sistema nervioso autónomo, el incremento del estrés oxidativo, la actividad del sistema renina-angiotensina-aldosterona pueden estar involucradas en su patogénesis 1.
Componentes del SM.
Obesidad.
La obesidad se origina por un balance energético positivo producto de la ingesta aumentada de alimentos. Este estado de acumulación lipídica necesita de la capacidad de adaptación por parte del tejido adiposo, incluyendo la formación de adipocitos nuevos, un proceso conocido como hiperplasia adipocítica.
No obstante, la capacidad del tejido adiposo para responder a las necesidades de acumulación de grasa tiene sus límites y si se sobrepasa puede sobrevenir la hipertrofia adipocítica y una respuesta inflamatoria.
La hipertrofia adipocítica origina adipocitos disfuncionales y una llegada y acumulación de macrófagos dentro del tejido adiposo. La infiltración de macrófagos dentro de este tejido, sumado a la presencia de adipocitos disfuncionales, lleva a una producción aumentada de adipoquinas proinflamatorias, que incluyen TNF-α (del inglés “Tumor necrosis factor alpha”), interleucina 6 (IL-6), inhibidor del activador del plasminógeno I (PAI-I) y ácidos grasos libres (AGL) entre otros 2.
Esto resulta en una inflamación crónica subclínica originalmente ubicada en el tejido adiposo que se propaga como una inflamación sistémica crónica.
En este proceso ocurre una acumulación ectópica de grasa en otros órganos o tejidos (hígado, músculo esquelético, corazón y páncreas entre otros). Como estos órganos no son capaces de acumular lípidos sin la alteración de sus funciones, sobreviene la lipotoxicidad en los mismos, que puede llevar finalmente a la resistencia a la insulina en dichos órganos 36.
Dislipidemia.
La dislipidemia del SM es la misma de la obesidad y de la diabetes tipo 2 y se caracteriza por niveles de triglicéridos (TAG) séricos aumentados, aumento de AGL, valores disminuidos de HDL-colesterol y un aumento de las partículas LDL pequeñas y densas 37,38.
La IR también se asocia fuertemente con este perfil dislipídico, pues esta condición causa una lipólisis aumentada a nivel de adipocito originando concentraciones elevadas de AGL. A su vez este aumento de los AGL causa una acumulación ectópica de grasa en el hígado, corazón, páncreas y músculo esquelético, entre otros. A nivel de hígado el influjo aumentado de AGL origina una producción y secreción aumentada de partículas de VLDL (lipoproteínas de muy baja densidad) originando de este modo el nivel elevado de TAG. Además, la IR ocasiona una menor actividad de la enzima lipoproteín-lipasa, disminuyendo el aclaramiento tanto de VLDL como de quilomicrones contribuyendo de forma adicional con la hipertrigliceridemia 2.
El estado de hipertrigliceridemia aumenta la actividad de la enzima CETP, intercambia TAG de las VLDL hacia las LDL y HDL en intercambio por colesterol esterificado de estas dos lipoproteínas. En este intercambio se forman partículas de HDL y LDL ricas en TAG, que son sustrato para la enzima lipasa hepática originando partículas de LDL pequeñas y densas y de HDL también pequeñas 39. El menor tamaño de las HDL aumenta su catabolismo ocasionando un menor nivel de HDL-colesterol. Las partículas LDL pequeñas y densas son altamente aterogénicas.
Hipertensión.
La hiperglicemia y la hiperinsulinemia activan el sistema renina-angiotensina (RAS, del inglés “renin angiotensina system”) pues incrementan la expresión de angiotensinógeno, de angiotensina II (ANG II) y del receptor AT1 y todo esto puede contribuir al desarrollo de la hipertensión, pues la ANG II ejerce varios efectos que modulan la presión sanguínea. Ambas condiciones, hiperglicemia e hiperinsulinemia, se presentan en la IR y en la obesidad 2.
La insulina tiene acciones sistémicas que afectan el sistema nervioso simpático, que participa en la regulación de la presión arterial y del riñón. La hipótesis de la insulina de la hipertensión propone que la hiperinsulinemia compensatoria que ocurre por efecto de la IR incrementa la reabsorción de sodio y la actividad simpática y ambos efectos combinados causan una elevación de la presión arterial 40.
Los adipocitos poseen la maquinaria enzimática involucrada en el RAS y de hecho sintetizan angiotensina II, y también aldosterona, y podría visualizarse a esta célula como un RAS en miniatura.
La ANG II puede reducir la utilización de la glucosa y la sensibilidad a la insulina, incrementar la IR en el músculo esquelético y en el tejido adiposo contribuyendo de este modo al SM 41.
Insulino resistencia.
El estado de insulino resistencia es un estado donde hay una menor respuesta por parte de los tejidos insulinodependientes a la presencia de la insulina.
En términos generales la génesis de la IR se ha asociado a la acumulación de lípidos en diferentes tejidos y órganos (hígado, músculo esquelético, páncreas y corazón entre otros) causada por la obesidad y por un estado inflamatorio crónico subclínico.
Esta acumulación de grasa causa lipotoxicidad dentro de la célula que trastorna el funcionamiento celular y puede llevar hasta la apoptosis. Este proceso de alteración o de estrés celular ocasiona una disfunción de algunas organelas, particularmente de las mitocondrias y del retículo endoplásmico 36.
Hiperglicemia.
La hiperglicemia en ausencia de diabetes puede resultar de un estado de IR, pues en condiciones normales el músculo esquelético es el principal tejido que capta la glucosa por acción de la insulina y en el estado de IR dicha captación no es la óptima, ocasionando un aumento de los niveles sanguíneos de glucosa.
Sumado a la anterior, el hígado, debido a la IR, produce y libera glucosa a la sangre, pues la insulina por el estado de IR a nivel hepático no logra inhibir la gluconeogénesis hepática (producción de glucosa).
La hiperglicemia es un factor de riesgo para diabetes tipo 2 por la presencia crónica de glucosa y de AGL aumentados.
La hiperglicemia está asociada con una secreción deteriorada de insulina, a causa de que los adultos con intolerancia a la glucosa han perdido cerca del 50% - 80% de su capacidad secretora de insulina. La función de las células β es mayor en individuos con prediabetes, comparado con individuos con SM y prediabetes 42. Esto apoya la noción que el SM deteriora la secreción de insulina.
La exposición crónica a nutrientes (glucosa y AGL) en concentraciones elevadas induce estrés oxidativo que puede causar disfunción de las células β y aún la muerte de dichas células 43. La diabetes tipo 2 (DT2) es el resultado de una IR crónica y una pérdida de la masa y de la función de las células β. La obesidad es el factor patogénico para el desarrollo de la IR, causando una acumulación intracelular lipídica en el páncreas. La IR crónica progresa a diabetes tipo 2 cuando las células β son incapaces de secretar cantidades adecuadas de insulina para compensar la sensibilidad disminuida a la misma, lo cual se debe principalmente a una disfunción secretora y a una pérdida significativa de células β funcionales 44.
Aunque la IR se observa en el estado de obesidad la mayoría de las personas obesas no desarrollan la enfermedad y la secreción aumentada de insulina, debido a una función aumentada de células β preexistentes o por expansión de la masa de células β, compensa y restaura los niveles de glucosa plasmática 45.
Los individuos obesos no diabéticos muestran un volumen aumentado relativo de células β en los islotes mientras los pacientes obesos y no obesos con una glucosa en ayunas alterada muestran una reducción de por lo menos del 40% en el volumen de las células β comparado con pacientes no diabéticos. La apoptosis está involucrada como el mecanismo primario causante de la disminución de la masa de células β en individuos diabéticos tipo 2. Conforme el número de células β por islote declina, el espacio de los islotes llega a ser dominado por la deposición de placas amiloides 46. Los factores llevando a un cambio en la función (expresión y secreción disminuida de la insulina) y en la masa de las células β son centrales a la patología de la DT2. Un decrecimiento tanto en la masa y en la función secretora de las células β es la característica común de la DT2.
La teoría prevaleciente para las causas de la falla de las células β durante la progresión a DT2 involucra la exposición crónica a la glucosa y a AGL, conocido como glucotoxicidad y lipotoxicidad, respectivamente 44.
Un flujo aumentado de glucosa y de AGL crea una carga tremenda sobre la oxidación mitocondrial llevando a una mayor producción de ROS (del inglés “reactive oxigen species”) y ocasionando un estrés oxidativo. Las células β tienen una capacidad limitada para enfrentar el estrés oxidativo, debido a un bajo nivel de enzimas antioxidantes 44.
Un nivel aumentado de ROS, o la presencia de estrés oxidativo, causa la activación de la vía de señalización de JNK que causa una secreción disminuida de insulina por medio de la traslocación nucleocitoplásmica del factor de transcripción PDX-1, un factor que se une al sitio promotor de la insulina e induce su expresión 47.
La disfunción y muerte de la célula β están conectados al estrés del retículo endoplásmico (ERE) y a la disfunción mitocondrial.
La IR exige una producción aumentada de insulina por parte de las células β y estas células responden incrementando su masa y la secreción de insulina. La producción de la insulina llega a representar hasta el 50% de la proteína total producida por la célula 48. Esto puede exceder la capacidad de plegamiento resultando en una acumulación de proinsulina no plegada en el lumen del retículo endoplásmico, generando el ERE. Una activación prolongada o excesiva de la UPR, mecanismo que se activa para tratar de normalizar al retículo endoplásmico, puede llevar a la apoptosis de células β por diferentes mecanismos. Alternativamente el ERE puede activar también vías inflamatorias que trastornan las funciones celulares y lleva a desórdenes metabólicos. También el ERE causa acumulación de ROS induciendo estrés oxidativo y el estrés oxidativo promueve el ERE, de modo que ambos elementos contribuyen de manera aditiva al desarrollo de desórdenes metabólicos 49. Cuando la UPR falla en su cometido de lograr la homeostasis del retículo endoplásmico y la función de esta organela se halla comprometida se induce la apoptosis celular.
La muerte de las células β por exposición a citoquinas proinflamatorias o por glucolipotoxicidad, fenómenos presentes en la obesidad y en el SM, ocurre a través de la vía mitocondrial intrínseca de apoptosis. En este mecanismo ocurre una permeabilización de la membrana mitocondrial externa y la liberación del citocromo c al citoplasma. Esta enzima en el citoplasma oligomeriza con el factor-1 activante de proteasa apoptótica (APAF-1, del inglés “apoptotic protease activating factor-1”) formando un apoptosoma, que activa a su vez a la caspasa 9. Esta caspasa activa a las caspasas 3 y 7 que ejecutan el programa de apoptosis 50.
Otra vía implicada en la disfunción y muerte de las células β es la NF-kB, que es activada por condiciones presentes en el SM, en la DT2 y en la obesidad. Esta vía promueve productos citotóxicos que exacerban la inflamación y el estrés oxidativo y promueven la apoptosis, llevando a disfunción celular o muerte celular respectivamente 49.
Estado protrombótico.
El estado protrombótico asociado al SM es el resultado de un grupo de alteraciones que involucran a las vías intrínseca y extrínseca de la coagulación, a la fibrinólisis y a la función plaquetaria, cada uno cooperando para favorecer una tendencia trombótica.
Hay niveles aumentados de los factores VIII, fibrinógeno, von Willebrand, y de Factor Tisular. Además, el número de micropartículas o microvesículas (MP) se encuentra aumentado. Las MP pueden contener factores de la coagulación, en particular Factor Tisular, y pueden contribuir a la amplificación de la respuesta trombótica 51.
El proceso de fibrinólisis está disminuido por un aumento de PAI-1 (4) y las plaquetas presentan un estado de activación caracterizado por la presencia de P-selectina, un marcador de activación plaquetaria 52.
La alteración del sistema fibrinolítico asociado a niveles aumentados de PAI-1 se considera que tiene un papel relevante en la tendencia protrombótica asociada al SM 16.
El riesgo trombótico está directamente correlacionado con el peso y una reducción de la IR periférica puede contribuir a reducir el riesgo trombótico en sujetos obesos 53.
Otros factores asociados al SM.
El SM tiene de fondo un estado inflamatorio crónico y bajo este hecho podría relacionarse con marcadores que detecten este estado inflamatorio.
La Proteína C Reactiva de alta sensibilidad (llamada de ahora en adelante PCR) es un marcador de inflamación de alta sensibilidad y se encuentra elevado en el SM. Además, hay una relación lineal entre el número de componentes del SM presentes y los niveles de PCR 54.
Ridker et al evaluaron la relación entre PCR y el SM entre mujeres sanas y encontraron que los niveles de PCR eran de 0.68, 1.09, 1.93, 3.01 y 5.75 mg/l para 0,1, 2, 3, 4 y 5 componentes del SM presentes 55,56.
Entonces a todos los niveles de severidad del SM la PCR adiciona información pronóstica sobre el riesgo subsecuente, pues los niveles de esta proteína contribuyen al riesgo cardiovascular aumentado 57.
La PCR no se limita a ser tan solo un marcador de riesgo cardiovascular, sino que también contribuye al daño vascular.
Esta proteína contribuye al daño endotelial, un evento que juega un papel crítico en la iniciación y la progresión de la enfermedad vascular aterosclerótica 58,59. La PCR deteriora la vasorreactividad del endotelio, disminuye la actividad de la eNOS (sintetasa del óxido nítrico endotelial) y la producción del NO, principal vasodilatador fisiológico. También contribuye al estrés oxidativo dentro de la célula endotelial 57.
Entonces hay una fuerte relación entre el nivel de la PCR y los componentes del SM y la adición de esta proteína puede ayudar a identificar a los pacientes en alto riesgo de diabetes tipo 2 y de enfermedad cardiovascular.
La prevalencia del SM aumenta también con los niveles séricos incrementados de ácido úrico (AU) y con una disminución de la tasa de filtración glomerular estimada 60.
Un riesgo aumentado en 1.6 veces para el SM fue observado en individuos con niveles de AU en el cuartil mayor comparado con el cuartil más bajo 61. En concordancia con lo anterior Yang et al encontraron un incremento en la incidencia del SM conforme aumentaba el valor del AU agrupado en terciles 62.
Tratamiento.
En esta sección se va a hablar del tratamiento en forma general y citando lo que la literatura indica y no pretende en ningún modo ser una guía para tratar el SM.
El SM es un estado inflamatorio crónico de bajo nivel con profundos efectos sistémicos. La identificación clínica y el manejo de los pacientes con SM es muy importante y el propósito del tratamiento es reducir las enfermedades subsecuentes asociadas a este síndrome.
El manejo efectivo incluye cambios en el estilo de vida, primariamente la pérdida de peso con dieta y ejercicio. El tratamiento farmacológico debe ser considerado para aquellas personas cuyos factores de riesgo no son reducidos adecuadamente con las medidas indicadas. El manejo clínico del SM es difícil porque no hay un método reconocido para prevenir o mejorar el síndrome como un todo. Entonces el tratamiento busca tratar cada componente del SM por separado, dando especial énfasis a aquellos componentes que son fácilmente manejables al tratamiento con drogas o fármacos.
Un primer paso es determinar el riesgo cardiovascular en los pacientes utilizando el algoritmo de Framingham para estimar el riesgo de ECV. La fórmula de Framingham incluye el fumado de cigarrillos, la presión arterial, el colesterol total, el HDL colesterol y la edad. Esta ecuación separa a los pacientes en tres categorías de riesgo basado en el riesgo a 10 años de enfermedad coronaria cardíaca: alto riesgo (el riesgo a 10 años es ≥ 20%), riesgo alto moderado (riesgo a 10 años entre 10% y 20%), riesgo moderado a bajo (riesgo a 10 años ≤ 10%) 63.
Reducción de peso. La obesidad es un importante factor de riesgo vascular y la pérdida de peso con medidas sobre el estilo de vida (dieta y ejercicio) continúa siendo la primera prioridad en pacientes con IR para controlar la dislipidemia y prevenir la DT2, pero es difícil lograrlo y mantenerlo. Por consiguiente las drogas que facilitan la pérdida de peso resultan útiles 39. Se ha recomendado una meta de reducción del peso del 10% en los primeros seis meses a un año y continuar perdiendo peso hasta llegar a un IMC menor de 25 2.
Dislipidemia. Para la dislipidemia la meta prioritaria es la disminución del LDL colesterol a menos de 130 mg/dl con la opción de disminuirlo a menos de 100 mg/dl en los individuos con alto riesgo y menos de 70 mg/dl en aquellos con muy alto riesgo. En pacientes con TAG ≥ 200 mg/dl el colesterol no HDL se convierte en una segunda meta. Después de alcanzar estas dos primeras metas la elevación del HDL colesterol se convierte en la tercera meta. En el caso del HDL colesterol no existen valores especificados por alcanzar, sino que este parámetro debe ser elevado todo lo que se pueda.
Las estatinas son consideradas como la droga más efectiva para disminuir el LDL colesterol, debido a sus mínimas interacciones droga-droga y a sus pocos efectos secundarios 63. La niacina se considera el mejor agente para elevar el HDL colesterol y para incrementar el tamaño de la partícula de HDL 64. La niacina también causa cambios positivos en la composición de las lipoproteínas, reduciendo la proporción de partículas de LDL pequeñas.
Hipertensión. La hipertensión debe ser tratada y las drogas antihipertensivas deben ser introducidas aun a presiones sanguíneas menores (≥130/≥80 mm Hg) en los pacientes con diabetes establecida. Las elevaciones moderadas de presión sanguínea pueden ser controladas con cambios en el estilo de vida. No obstante, si la hipertensión no puede ser controlada con estas terapias de estilos de vida, deben utilizarse drogas antihipertensivas para prevenir los efectos adversos a largo plazo, como el infarto agudo de miocardio, el derrame cerebral y la enfermedad renal crónica 65.
En el 2015 un grupo de trabajo, The Cardiometabolic Think Tank, revisó el tema del SM y emitió un modelo de atención para las personas con SM. El modelo reconoce la heterogeneidad del SM y la necesidad de estrategias individualizadas de cuidado. El modelo establece cuatro estados (A, B C y D) donde ubicar a cada paciente. Elprimer estado, A, reconoce a los pacientes sin SM y que no cumplen, o tienen, ningún criterio del mismo; el estado B comprende a los pacientes sin SM, pero con más de un criterio; el estado C ubica pacientes con SM pero sin daño en órganos y el estado D, comprende a los pacientes con SM y con daño terminal en órganos. Para cada estado el grupo estableció una terapia particular y según se avanza de estado la terapia se vuelve cada vez más exhaustiva. El modelo se mueve hacia estados cada vez más severos sobre la base de criterios diagnósticos y factores de riesgo establecidos y marcadores de riesgo residual. Cada estado propone estrategias terapéuticas más intensivas para tratar el SM y sus factores de riesgo 5.
El grupo de trabajo por supuesto enfatiza en la prevención del SM y en el manejo del paciente dentro de un modelo que contemple la salud individual, comunitaria y pública.
Conclusiones.
El SM abarca a un grupo de componentes estrechamente relacionados entre sí, que incrementan el riesgo de ECV y de diabetes mellitus tipo 2: obesidad abdominal, dislipidemia, hiperglicemia, insulino resistencia e hipertensión.
Existen diferentes criterios o definiciones para establecer la presencia o ausencia del SM en los individuos.
Un tejido adiposo disfuncional por efecto de la hipertrofia patológica de los adipocitos, producto de una ingesta calórica aumentada, presenta una secreción desbalanceada de adipoquinas y citoquinas, con un predominio de factores proinflamatorios, dando origen a un estado inflamatorio crónico de bajo nivel o subclínico. Dentro del marco de este proceso inflamatorio crónico se desarrolla el SM. Aparte de la obesidad, la resistencia a la insulina es crucial para la génesis del SM.
Hay un efecto aditivo de los componentes del SM para el riesgo de desarrollar ECV y diabetes mellitus tipo 2 y para desarrollar otras enfermedades metabólicas.
El tratamiento del SM incluye en principio cambios en el estilo de vida enfatizando en la reducción de peso y la práctica del ejercicio. El tratamiento farmacológico se incluye también como complemento a las otras medidas citadas.

