











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El término muerte súbita tiene definiciones similares pero no unificadas que varían discretamente en razón del área de la medicina o incluso del país donde se defina. Desde el punto de vista médico legal, la muerte súbita se define como un evento fatal e inesperado que ocurre en un individuo aparentemente sano1. La Organización Mundial de la Salud agrega un factor cronológico indicando que es aquella que ocurre en las primeras 24 horas de acontecidos los síntomas.2
En la literatura médica costarricense, se considera muerte súbita, como aquel deceso de causa no violenta que sucede sorpresiva y rápidamente a un individuo en aparente buen estado de salud previo, o portador de una patología aguda o crónica cuyo desenlace fatal era altamente improbable3
Entre las principales causas se encuentran las de origen cardiovascular. Su incidencia es difícil de determinar y varía de un país a otro4. En Estados Unidos se estima que ocurren alrededor de 350 000 muertes súbitas de origen cardíaco anuales1, sin embargo, en Costa Rica este dato no se encuentra documentado de manera que permita establecer de forma precisa la incidencia.
Son varias las etiologías que pueden causar muerte súbita cardíaca, entre las principales se encuentran la cardiopatía isquémica, la cardiopatía hipertrófica, valvulopatías, cardiopatías congénitas y otros trastornos cardíacos menos comunes como las anormalidades electrofisiológicas primarias5. Entre este último grupo destacan el síndrome de Wolf Parkinson White, el síndrome QT prolongado congénito, bloqueos atrio ventriculares y el síndrome de Brugada, el cual se define como una canalopatía autosómica dominante que afecta canales de sodio6.
Precisamente, las causas genéticas más comunes de arritmias ventriculares malignas y muerte súbita cardíaca en población joven sin patología estructural conocida, son el síndrome de QT prolongado y el Síndrome de Brugada7. En esta ocasión se realiza una revisión, con especial énfasis en la genética, fisiopatología, abordaje diagnóstico y manejo para prevención de muerte súbita del Síndrome de Brugada.
Definición y epidemiología
El síndrome de Brugada se encuentra dentro del grupo de enfermedades cardíacas conocidas como canalopatías8. En este grupo de enfermedades, la patogenia se debe una alteración en los canales iónicos transmembrana responsables del potencial de acción del miocito. La consecuencia más fatal de las canalopatías, es la generación de arritmias ventriculares, que a su vez predisponen a muerte súbita.
De manera más específica, se define el síndrome de Brugada como una canalopatía que afecta canales de sodio, producto de una variante genética, principalmente de herencia autosómica dominante con penetrancia variable6.
Aunque cabe destacar, que en un importante número de casos puede aparecer de forma esporádica sin que se encuentre presente en otros familiares6,8.
Esta variante genética produce una disminución en el flujo de iones de sodio, que en condiciones normales propician una generación adecuada del potencial de acción del miocito. El resultado final puede ser una repolarización heterogénea ventricular, que a su vez puede causar arritmias ventriculares maligas6.
Se estima que tiene una prevalencia de 1 a 5/10 000 habitantes, y que es responsable de hasta un 4-12% de todas las muertes súbitas y de un 20% de las muertes súbitas cardíacas acontecidas en individuos sin patología estructural conocida9. Es más frecuente en el género masculino y la edad media de diagnóstico es a los 42 años9.
Historia
Han pasado 25 años desde que Pedro y Josep Brugada lo describieron por primera vez en 199211. En las primeras caracterizaciones se describió como un síndrome clínico con imágenes electrocardiográficas de bloqueo de rama derecha del Haz de His y elevación persistente del segmento ST en derivaciones V1, V2 y V3, que además era causante de arritmias ventriculares y muerte súbita en pacientes sin cardiopatía estructural evidente, esa primera descripción incluía 8 pacientes10,11,13. Aunque el término síndrome de Brugada se acuñó por investigadores japoneses en 1996, posterior a la publicación de esa primera serie de casos, se cuestionó la existencia de la entidad como un nuevo síndrome12. Sin embargo, en 1998 se descubre el defecto genético responsable del síndrome de Brugada, poniendo fin a la discusión12. Posteriormente se fueron reportando más series de casos que han permitido la elaboración de estudios en los cuales se ha logrado definir de forma detallada la fisiopatología y los aspectos genéticos y moleculares de la enfermedad que conocemos en la actualidad8.
Genética
Con la conclusión el proyecto del genoma humano en el 2003, se ha estimado que nuestro genoma contiene alrededor de 30 000 genes que a su vez expresan aproximadamente 100 000 proteínas13. A raíz de esto, se han desarrollado muchos métodos de secuenciación de ADN, uno de los primeros fue la secuenciación de Sanger, que fue el que se utilizó en 1998 para identificar la alteración genética que produce el síndrome de Brugada13.
Se determinó que la mutación del gen SCN5A, que codifica la subunidad alfa del canal de sodio cardíaco voltaje dependiente Nav1.5 se asociaba con el síndrome14. Sin embargo, con los avances tecnológicos en genómica, se han reportado varios genes asociados, de los cuales la mayoría codifica para canales de sodio y algunos para canales de potasio y calcio, reconociéndose así varios subtipos genéticos del síndrome de Brugada13,14.
A pesar de esto, el gen SCN5A continúa siendo el más asociado con esta patología, habiéndose reportado más de 300 mutaciones del mismo en pacientes con diagnóstico confirmado. Se ha estimado que la alteración en este gen es responsable del 15-25% de los casos, sin embargo hasta en un 70% de los pacientes clínicamente y electrocardiográficamente diagnosticados con el síndrome, se desconoce la alteración genética causante13, es decir, actualmente se sabe que la mayoría de casos de síndrome de Brugada no tienen alteración genética conocida15.
En cuanto a la penetrancia, que es la proporción de individuos con la mutación que manifiestan los signos y síntomas clínicos, se ha visto que en el síndrome de Brugada es variable. En un estudio realizado en familias portadoras de la mutación en el gen SCN5A se vio una penetrancia del 12,5-50%16. Además, la penetrancia se relaciona con el sexo y la edad, ya que los eventos más letales ocurren en el género masculino y después de los 40 años17. A pesar de que la transmisión genética es igual para ambos géneros, el fenotipo de Brugada es de 8 a 10 veces más prevalente en hombres con respecto a mujeres7.
Asimismo, el riesgo relativo de muerte súbita es 3,3 veces mayor en el género masculino, esto puede deberse al efecto de hormonas masculinas en las corrientes de canales iónicos cardíacos, sin embargo el mecanismo no se ha dilucidado de manera precisa18.
Presentación clínica
La forma más típica de presentación es síncope o muerte súbita resucitada debido a taquicardia ventricular polimórfica o fibrilación venticular6. Usualmente los síntomas se presentan durante el reposo o durante el sueño, sin embargo se ha visto que en población europea por lo general se presenta en cualquier otra condición que no sea reposo19.
Un gran número de pacientes tienen antecedente de muerte súbita inexplicada en familiares20.
En niños generalmente se presenta como una taquicardia ventricular monomórfica, que en la mayoría de los casos es desencadenada por fiebre6.
Fisiopatología
Durante las fases 0 y 1 del potencial de acción miocárdico normal, la corriente de entrada de sodio y la corriente de salida transitoria de potasio, conocida como ITo, causan la morfología de espiga de la repolarización temprana y posteriormente junto con corrientes de calcio continúa la fase 2 o meseta6. En el síndrome de Brugada esa corriente de entrada de sodio se ve disminuida, y la corriente de potasio y otras corrientes de calcio continúan de forma normal.
Esto ocurre en el epicardio, mientras que el endocardio se mantiene sin alteraciones, lo que crea un gradiente de voltaje transmural. El efecto de esto, es que aumente la incisura o muesca de la fase 1 en el epicardio del ventrículo derecho. En el electrocardiograma esto se traduce en una elevación del punto J y la elevación del ST característica de Brugada6,21.
Las arritmias se producen debido a la repolarización heterogénea en diferentes sectores del epicardio ventricular producida por ese gradiente de voltaje transmural, lo que a su vez provoca un fenómeno de reentrada con el subsecuente desarrollo de extrasístoles que pueden terminar en taquicardia o fibrilación ventricular6,21.
Diagnóstico
La sospecha diagnóstica inicial surge mediante historia clínica y un patrón electrocardiográfico específico. Los criterios diagnósticos electrocardiográficos incluyen elevación del segmento ST en V1, V2 y V3, dentro del cual se reconocen dos patrones: el tipo I, caracterizado por una elevación descendente y cóncava o rectilínea del segmento ST ≥ 2 mm en más de una derivación precordial derecha (V1-V3), seguida de ondas T negativas simétricas. El patrón tipo II, es caracterizado por un onda r’ de ≥ 2mm con respecto a la línea isoeléctrica que es seguida por una elevación del ST convexa y una onda T positiva o aplanada en V2 y onda T de morfología variable en V121 (fig 1).
Ante la presencia de esos patrones en el ECG, se debe cumplir uno o más de los siguientes criterios clínicos para realizar el diagnóstico21:
a. Paciente sobreviviente a paro cardíaco.
b. Presencia de taquicardia ventricular polimórfica.
c. Antecedente de síncope no vaso vagal.
d. Antecedente de muerte súbita en familiares menores de 45 años sin síndrome coronario agudo.
e. Patrón electrocardiográfico tipo I en familiares.
Además, es importante mencionar que esos cambios electrocardiográficos se pueden presentar de forma espontánea o ante la administración de antiarrítmicos de la clase I (ajmalina, flecainida, pilsicainida)6. A la administración de estos antiarrítmicos se le conoce como test de provocación y está indicado en pacientes que tienen un electrocardiograma no diagnóstico para síndrome de Brugada y en los que se deba excluir el síndrome por antecedente de paro cardíaco sin patología estructural conocida o por la presencia del patrón electrocardiográfico de Brugada en familiares6.
Si bien es cierto los cambios electrocardiográficos mencionados son la base diagnóstica, son cambios que pueden ser dinámicos o intermitentes o que se pueden presentar en otras condiciones21 como las que se presentan en el siguiente apartado.
Se ha visto en biopsias de pacientes con síndrome de Brugada típico, una fibrosis y pérdida de las uniones adherentes en todo el tracto de salida del ventrículo derecho22.
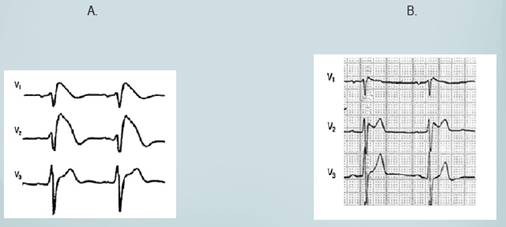
Figura 1 Patrones electrocardiográficos en Síndrome de Brugada. A) Patrón tipo I; elevación descendente y cóncava del segmento ST ≥ 2 mm en más de una derivación precordial derecha (V1-V3), seguida de ondas T negativas simétricas y B) patrón tipo II; onda r´ de ≥ 2mm con respecto a la línea isoeléctrica que es seguida por una elevación del ST convexa y una onda T positiva o en V2 y onda T de morfología variable en V1.
Diagnóstico diferencial
Como se mencionó, es importante destacar que muchas condiciones clínicas pueden producir elevación del segmento ST en derivaciones precordiales derechas, imitando el patrón de Brugada13. Se han mencionado 6 categorías etiológicas que imitan dicho patrón, las cuales son23:
1. Condiciones metabólicas y toxicológicas; entre las principales están exposición a ciertos tipos de drogas como;
cannabis, cocaína, heroína y ketamina, además desbalances hidroelectrolíticos como hiper o hipokalemia, hipercalcemia y alteraciones metabólicas como hipotermia e insuficiencia adrenal.
2. Compresiones mecánicas extrínsecas como pectum excavatum o tumores mediastinales13.
3. Infarto de ventrículo derecho y embolismo pulmonar.
4. Enfermedad pericárdica y miocárdica.
5. Mala calibración electrocardiográfica o uso de filtros inadecuados.
6. Causas misceláneas.
A todas estas condiciones se les conoce como Síndrome de Brugada adquirido o fenocopia de Brugada13. La diferenciación entre este último y el Brugada congénito se debe hacer reconociendo la presencia o no de factores desencadenantes como los mencionados. Una vez corregido el factor identificado, si el patrón electrocardiográfico revierte y no se desencadena con el test de antiarrítmicos clase I, se dice que se trataba de un patrón de Brugada adquirido. Caso contrario, si no revierte o bien, si revierte pero se desencadena con los antiarrítmicos, se trataba de un síndrome de Brugada congénito13 .
Manejo y tratamiento
Se ha determinado que el desfibrilador automático implantable (DAI), es la principal herramienta para la prevención de muerte súbita en pacientes con esta patología24,25,26. Sin embargo es indispensable estratificar el riesgo e identificar de forma precisa cuales pacientes se benefician de dicho dispositivo.
En un estudio realizado en Francia, Italia, Países Bajos y Alemania, conocido como FINGER, se determinó que los únicos factores independientes de riesgo arritmogénico en pacientes con síndrome de Brugada son; paro cardíaco previo, síncope y patrón electrocardiográfico tipo I (fig 1)24. Otros estudios sugieren que los pacientes de más alto riesgo de muerte súbita, son aquellos que presentan un patrón electrocardiográfico tipo I y con al menos otros dos factores de riesgo, entre los que se incluyen síncope, familiares con patrón de Brugada o estudios electrofisiológicos positivos25.
De forma específica, se dice que en síndrome de Brugada las indicaciones para DAI son; pacientes sobrevivientes a paro cardíaco, y pacientes con el patrón electrocardiográfico y antecedente de síncopes, taquicardia o fibrilación ventricular, debido al alto riesgo de eventos recurrentes6,24. Además se ha visto que el antecedente de muerte súbita en familiares no es un factor predictor ni aumenta el riesgo de muerte súbita en este grupo de pacientes6 .
La implantación del DAI tiene una alta tasa de complicaciones, y en general no está recomendado para pacientes asintomáticos. Entre las principales complicaciones se incluyen descargas innecesarias e infecciones en el sitio del dispositivo25. Por lo tanto es importante recalcar la necesidad de realizar, previo a la colocación del dispositivo, una adecuada estratificación del riesgo.
Dada la alta posibilidad de arritmias ventriculares recurrentes y la subsecuente descarga del dispositivo en pacientes adecuadamente estratificados, y el impacto en la calidad de vida que esto pueda tener, se ha intentado encontrar alternativas farmacológicas. La quinidina es un antiarrítmico de la clase Ia que inhibe la corriente de potasio Ito, y se ha sugerido como tratamiento adicional en estos pacientes26,27. En un estudio realizado en España se vio una reducción promedio por paciente en las descargas del dispositivo de 203 a 41 tras el uso de quinidina en un período promedio de 60 meses. Concluyéndose que el uso de quinidina disminuye la recurrencia de arritmias ventriculares y reduce el número de descargas eléctricas del DAI, además se vio que la discontinuación del tratamiento aumentaba la recurrencia26. Otros autores recomiendan el uso de quinidina en pacientes asintomáticos, dado que se desconoce el porcentaje de pacientes nacidos con síndrome de Brugada que desarrollarán arritmias ventriculares y dadas las complicaciones de implantación del DAI de forma generalizada y profiláctica, especialmente en población joven27.
Por último, es importante mencionar que en la mayoría de muertes súbitas con desenlace fatal, nunca llega a conocerse la etiología desencadenante. No existen análisis en autopsias que permitan determinar el padecimiento de canalopatías sin alteraciones estructurales, como lo son el síndrome de QT prolongado congénito y el síndrome de Brugada, por lo tanto, en autopsias solo se puede especular que la causa de muerte súbita haya sido una arritmia ventricular. Ante este hecho, se ha recomendado la realización de autopsias moleculares y estudios genéticos en familiares de primer y segundo grado de las víctimas, ya que esto permitiría no solo identificar la causa de muerte, sino identificar familiares en riesgo de sufrir eventos similares28.

