










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina Legal de Costa Rica
On-line version ISSN 2215-5287Print version ISSN 1409-0015
Med. leg. Costa Rica vol.31 n.2 Heredia Sep./Dec. 2014
Reporte de un caso
Carcinoma de Merkel (carcinoma neuroendocrino)
Pedro Madriz de Haan, Greivin Rodríguez*+
Resumen:
Desde que el carcinoma de Merkel fue descrito por primera vez, hace poco más de cuatro décadas por Cyril Toker en el año de 1972; ha despertado el interés de los colegas médicos. Factores de esta patología como: su célula de origen, etiología, evolución y tratamiento al día de hoy se mantienen en discusión y se modifican con el paso del tiempo. El advenimiento de la microscopía electrónica y la inmunohistoquímica han colaborado en la definición etiológica de esta neoplasia y otros descubrimientos como el del poliomavirus de células de Merkel, han brindado información importante sobre la génesis de este proceso. El tratamiento del carcinoma de células de Merkel sigue sufriendo leves variaciones con el paso del tiempo y se mantiene en discusión a la fecha. Presentamos el caso de una paciente con un carcinoma de células de Merkel con revisión de la literatura actual.
Palabras clave:
Carcinoma de Merkel, dermatopatología, anatomía patológica, célula de Merkel, inmunohistoquímica, microscopía electrónica
Abstract:
Since Merkel carcinoma was first described more than four decades ago by Cyril Toker little in the year 1972; has attracted interest from medical colleagues. Factors of this disease as their cell of origin, etiology, course and treatment today remain under discussion and change over time. The advent of electron microscopy and immunohistochemistry have collaborated in the etiological definition of this neoplasm and other discoveries such as the Merkel cell polyomavirus, have provided important information on the genesis of this process. The treatment of Merkel cell carcinoma is still suffering slight variations over time and remains in discussion to date. We report a patient with Merkel cell carcinoma with review of the current literature.
Key words:
Merkel cell carcinoma, dermatopathology, surgical pathology, Merkel cell, immunohistochemistry, electron microscopy
Introducción
El carcinoma de células de Merkel (CCM) de la piel es también conocido como carcinoma trabecular, carcinoma cutáneo neuroendocrino o Merkeloma. Es una neoplasia primaria cutánea poco común altamente maligna. Se cree que se origina en las células de Merkel, que están localizadas en la membrana basal de la epidermis y folículos pilosos, asociados con las terminaciones nerviosas sensitivas en las papilas dérmicas, específicamente los mecanoreceptores (P. Tai, 2013).
La célula de Merkel:
Gracias a la microscopía electrónica se ha obtenido conocimiento significativo de la anatomía celular y esto ha contribuido en el caso de la célula de Merkel, se demostró que ésta son ovales y claras con una medida de 10-15 micras en el eje de mayor longitud, con un núcleo lobulado y gránulos en su citplasma que muestran positividad para tinciones de citoqueratinas y neurofilamentos (B.L. Munger, 1965). Además estas células tienen protrusiones en forma de picos que les permiten entremezclarse con los queratinocitos circundantes. Las terminaciones nerviosas se encuentran llenas de mitocondrias y vesículas claras (B.L. Munger, 1965).
Uno de los datos más interesantes en cuanto al perfil de expresión de proteínas, es que los marcadores epiteliales como las citoqueratinas y los marcadores neuroendocrinos como la enolasa neurono específica, sinaptofisina y cromogranina pueden ser positivos en las células de Merkel. En particular la citoqueratina 20 es de un valor importante como altamente específico en las células de Merkel en el epitelio escamoso normal (Z. Halata, M. Grim, K.I., Bauman, 2003).
Carcinoma de células de Merkel:
El CCM fue descrito por primera vez en el año de 1972 por Cyril Toker, como un carcinoma trabecular de la piel, sugiriendo su origen de las células de las glándulas sudoríparas, aunque posteriormente gracias a la microscopía electrónica y a la demostración de gránulos neurosecretores en las células que los conformaban, se determinó su relación con las células de Merkel (C. K. Tang, C. Toker, 1978). Los análisis histoquímicos e histogenéticos acuñaron el término “carcinoma neuroendrocrino de la piel” para esta neoplasia, colocándolo en la familia de los APUDomas (R. K. Sibley, J. Rosai, E. Foucar, 1980) aunque el origen del tumor de una célula de Merkel no ha sido establecido en forma definitiva.
El CCM ocurre en predominante en pacientes mayores de raza blanca, con una incidencia semejante en hombres y en mujeres. El 78% de los pacientes son mayores de 59 años, aunque en algunas publicaciones se ha mencionado una predilección leve hacia el sexo femenino (A. B. Akosa, D.V. Nield, M. N. Saad, 1994). No existe predilección por sexo en el grupo de pacientes menores de 60 años, después de los 60 años se observa con más frecuencia en pacientes femeninas.
Esta neoplasia se encuentra con mayor frecuencia en la cabeza y en el cuello (50.8%), o en las extremidades (33.7%). La etiología exacta no se conoce pero se ha postulado que puede estar relacionado con la exposición solar, inmunosupresión y la infección por un poliomavirus; el descubrimiento del poliomavirus de la célula de Merkel ha sido un gran avance en el entendimiento de esta enfermedad (H. Feng, M. Shuda, Y. Chang, P. S. Moore, 2008). Esta neoplasia se observa con mayor frecuencia en pacientes con inmunosupresión ya sea, pacientes trasplantados o pacientes con HIV.
La asociación de esta entidad con el poliomavirus puede explicar el aumento en su incidencia en pacientes con inmunosupresión. El aspecto inmunológico de esta neoplasia es bastante interesante, de hecho existen reportes de casos con remisión completa o parcial (H. Takenaka, et al., 1997).
La incidencia de esta neoplasia ha aumentado y esto puede deberse a un aumento en la detección de la enfermedad o al envejecimiento de la población en general. Existen datos de países europeos como Holanda y Finlandia donde se encuentra que la incidencia va desde
Debido a lo poco frecuente que es esta enfermedad la mayoría de los cirujanos, no tienen mucha experiencia en el manejo de la misma. Existen algunas controversias en la literatura en cuanto a las opciones terapéuticas, especialmente desde el punto de vista adyuvante. Los tratamientos apropiados en la enfermedad recurrente y metastásica de esta enfermedad se vuelven un dilema debido a su incidencia en los pacientes adultos mayores, cuando la tolerancia al tratamiento agresivo no es buena (P. Tai, 2013).
Resumen de caso:
El caso en cuestión corresponde a una paciente femenina de 77 años de edad, quien es ama de casa. Dentro de los antecedentes personales no patológicos no presenta datos de importancia y en sus antecedentes personales patológicos se encuentra un glaucoma, con el antecedente de un trasplante de córnea.
La historia del padecimiento de la paciente inicia tres meses previos a la consulta con un dermatólogo y refiere una masa de crecimiento lento en su mejilla derecha, esta masa no era visible en la piel y era palpable en el tejido celular subcutáneo, midiendo
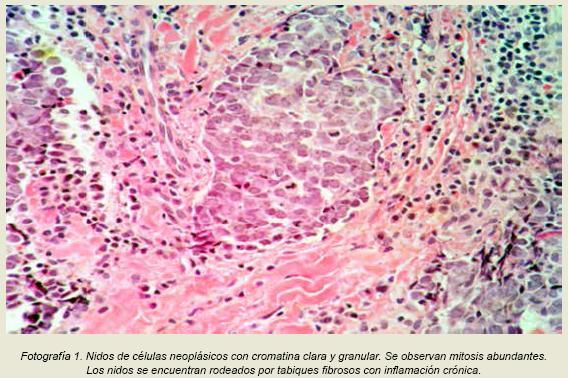
Piel con epitelio sin cambios, a nivel dérmico y subcutáneo se observaron brotes tumorales constituidos por células con escaso citoplasma y núcleos con cromatina dispersa y pequeños nucléolos con figuras de mitosis. Esos nidos estaban rodeados de abundante inflamación crónica (Fotografía 1).
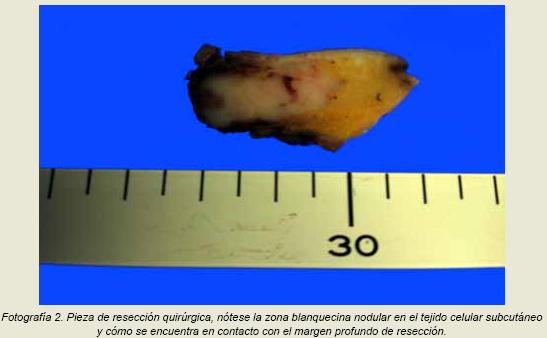
El patólogo a cargo de analizar esta biopsia mencionó que la neoplasia era maligna y para determinar la estirpe de la misma recomendó realizar tinciones de inmunohistoquímica, aunque recalcó que probablemente se trataba de una neoplasia neuroendocrina. Debido a esto a la paciente se le realizó una cirugía con la intensión de resecar la lesión en su totalidad quince días después de la biopsia diagnóstica. La resección se realizó sin complicaciones y se obtuvo un fragmento de piel y tejido adiposo de
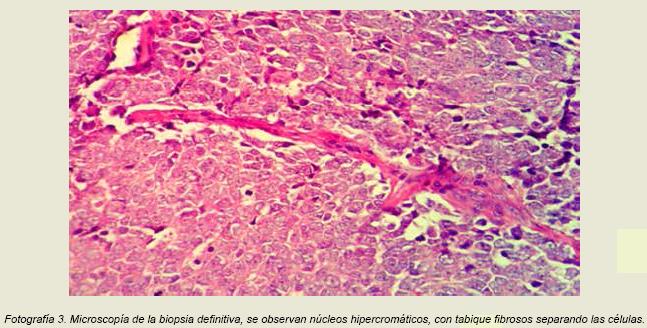
En el examen histológico se describen núcleos hipercromáticos y citoplasma mal definido, las células neoplásicas se disponen en lóbulos que se encuentran divididos por finos tabiques de tejido fibroso en patrón organoide (fotografía 3). La neoplasia se encontraba en contacto con el margen profundo de resección.
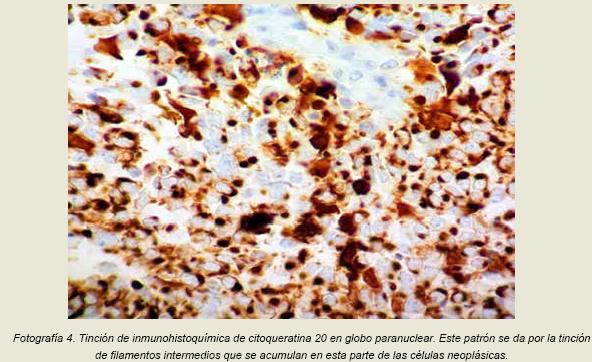
A esta muestra se le realizaron tinciones de inmunohistoquímica para confirmar la estirpe que se había sugerido anteriormente, dentro de las que se realizaron se encontraba la cromogranina y la sinaptofisina, ambas para determinar el origen neuroendocrino de las células neoplásicas y en ambos casos las células fueron positivas para estos marcadores; también se realizó pancitoqueratina, citoqueratina 20 y citoqueratina 7; que fueron positivas en las células neoplásicas en un patrón característico conocido como “globo paranuclear”(fotografía 4), además de un ki67 que fue positivo en un 80% de las células neoplásicas y un TTF-1 que fue negativo.
Discusión:
Los pacientes con carcinoma de Merkel requieren un diagnóstico temprano y un planteamiento en cuanto a conducta de tratamiento se refiere, una alternativa diagnóstica que se ha tomado en cuenta últimamente es la biopsia por aspiración con aguja fina (BAAF), ya que facilita el diagnóstico temprano y ayuda a planear la cirugía correspondiente (P. Tai, 2013). La BAAF se puede realizar en pacientes adultos mayores en comparación con la biopsia quirúrgica y teñida con tinción de Papanicolaou muestra: celularidad aumentada, grupos no cohesivos de células de pequeñas a medianas con núcleos uniformes redondondos u ovales, con moldeamiento, cromatina fina, micronucleolos y escaso citoplasma (K. T. Ostovic, et al., 2010). Ya que este tumor tiene un alto potencial de tener recurrencia local, así como diseminación linfática y en forma frecuente se observa en relación con otros tumores, se recomienda hacer varias BAAF de diferentes lesiones en el mismo paciente (H. Zhang, G. Gupta, X. Y. Yang et al., 2009).
Desde el punto de vista histológico el diagnóstico del CCM se debe realizar por medio de los hallazgos en la hematoxilina eosina y, de forma aún más importante en la inmunohistoquímica (positividad de marcadores epiteliales como las citoqueratinas con la tinción clásica en globo paranuclear y de marcadores neuroendocrinos). La neoplasia frecuentemente se encuentra en la dermis y se extiende hacia el tejido celular subcutáneo, la epidermis se encuentra comprometida de forma infrecuente y rara vez se observa ulceración.
El tumor puede estar compuesto por células aisladas, sábanas discohesivas y estructuras semejantes a rosetas; las células neoplásicas son ovoides y muy uniformes, la cromatina es granular fina y con figuras de mitosis frecuentes y se han descrito tres patrones: (1) sólido – el más común, compuesto por grupos irregulares de células tumorales conectados por bandas de tejido conectivo, (2) trabecular – cordones bien definidos de células que forman infiltran en forma difusa y (3) difuso – se observa pobre cohesión celular y un patrón de crecimiento difuso linfoma-like (P. Tai, 2013).
En la inmunohistoquímica es característico encontrar globos paranucleares con las tinciones de citoqueratinas y neurofilamentos. El hallazgo patonogmónico de esta neoplasia se encuentra en la microscopía electrónica y son los gránulos neurosecretores con un diámetro de 100-250nm rodeados por remolinos de filamentos intermedios, además de procesos digitiformes de 0.1-0.25 micras de diámetro se han descrito también (Y. Yamashita, K. Toida, and H. Ogawa, 1993).
En el año 2008 Feng y colaboradores encontraron secuencias virales en 4 muestras de carcinoma de Merkel, posterior a un análisis de secuencia se mostró se trataba de un poliomavirus, que posteriormente se llamó poliomavirus de células de Merkel. Posteriormente se encontró una prevalencia de un 40% de este virus en los carcinomas (M. Agelli, 2010). En particular los poliomavirus codifican por antígenos T grandes y pequeños que se unen a las proteínas del huésped, facilitando: replicación viral, inactivando proteínas supresoras de tumores como p53 y Feng con sus colegas demostraron el mismo patrón de integración viral monoclonal en el 50% de las muestras que examinaron; siendo el mismo patrón en el tumor primario como en la metástasis, indicando que la integración viral monoclonal precedió la diseminación de la neoplasia (H. Feng, 2008). Esto brinda implicaciones terapéuticas, ya que la expresión de la proteína grande T del poliomavirus de células de Merkel, se sobre-expresa en el tumor primario y ganglios linfáticos y se puede usar como objetivo terapéutico en pacientes con enfermedad diseminada y del mal pronóstico (Q. Zeng, 2012).
En cuanto al manejo de estos pacientes con esta patología el manejo de primera elección y sobre el cual existen dos estudios (uno retrospectivo y otro prospectivo) (I. Erovic y B. Erovic, 2013), es la cirugía con radioterapia adyuvante; ya que ha demostrado aumento en la sobrevivencia libre de enfermedad loco-regional. Aunque no se demostró aumento en la sobrevida en general (M. Poulsen, et al., 2013).
La cirugía de Mohs, se introdujo como método para el tratamiento de entidades neoplásicas en 1930, particularmente en sitios delicados, se han realizado algunos estudios con esta técnica en pacientes con CCM donde se ha demostrado que la misma es confiable y costo efectiva (J. D. Boyer, et al., 2002), en estos estudios se demostró que no existe diferencia significativa en la sobrevida libre de enfermedad cuando se compara la cirugía de Mohs contra resección quirúrgica con radioterapia, aunque esta última si disminuyó la incidencia de recurrencia local y de metástasis en tránsito en compraración a la cirugía de Mohs (J. D. Boyer, et al., 2002). El CCM es altamente radiosensible y en ocasiones en donde no se puede realizar la cirugía, la radioterapia por sí misma, ofrece excelentes resultados y control regional (B. D. Lawenda, et al., 2008).
En el caso de la quimioterapia, lamentablemente no existe tratamiento de primera línea para el CCM, de hecho la quimioterapia se utiliza únicamente en pacientes con enfermedad avanzada o en pacientes con enfermedad recurrente, no resecable o diseminada. Por estas razones el resultado de la terapia es sumamente controversial e inclusive se ha reportado menor sobrevida para los pacientes que recibieron quimioterapia versus los que no la recibieron (P. J. Allen, et al., 2005).
Conclusión:
Desde el descubrimiento de la célula de Merkel en el siglo 19 y con la posterior descripción de una neoplasia conformada por este tipo de células se ha realizado un gran avance en el diagnóstico, tratamiento y seguimiento de los pacientes que presentan esta patología. Con la descripción por microscopía electrónica de los gránulos en el citoplasma de las células de Merkel y su correlación con la tinción por inmunohistoquímica con citoqueratina 20 característica en globo paranuclear, se ha podido realizar un diagnóstico preciso, así como una valoración de la patología incluyendo factores como su evolución, riesgo de metástasis y recurrencia.
Otro parámetro importante en la evolución del manejo y entendimiento de esta patología es el descubrimiento de su relación con el poliomavirus, de manera que inclusive se le llama poliomavirus de células de Merkel; el cuál como podemos ver en esta revisión implica un comportamiento biológico diferente en los pacientes que tienen neoplasias con el virus y los que no. Esto, como todo hallazgo importante brinda una gama de oportunidades diferentes al paciente y permite una estratificación del riesgo dependiendo de las características de su enfermedad en particular, una “individualización” del CCM.
Existen varios avances en cuanto al tratamiento de este tipo de neoplasia dentro de los que se encuentran la realización de varios estudios donde se analizan las conductas terapéuticas a seguir como: cirugía, radioterapia, cirugía de Mohs y quimioterapia (este último con los resultados menos esperanzadores).
De todas formas es importante conocer las opciones para ofrecer a cada paciente y dependiendo de las características intrínsecas de la neoplasia así como del paciente en particular se pueda curar o prolongar la sobrevida libre de enfermedad.
Bibliografía:
1. Tai P,“A Practical Update of Surgical Management of Merkel Cell Carcinoma of the Skin”, Surgery Volume 2013, Article ID 850797, 17 pages. [ Links ]
2. B. L.Munger, “The intraepidermal innervation of the snout skinof the opossum. A light and electron microscope study, with observations on the nature of Merkel’sTastzellen,” Journal of CellBiology, vol. 26, no. 1, pp. 79–97, 1965. [ Links ]
3. Z. Halata, M. Grim, and K. I. Bauman, “Friedrich Sigmund Merkel and his “Merkel cell”, morphology, development, and physiology: review and new results,” Anatomical Record Part A, vol. 271, no. 1, pp.
5. R. K. Sibley, J. Rosai, and
8. H. Takenaka,
9. B. A. Reichgelt and O. Visser, “Epidemiology and survival of Merkel cell carcinoma in the
10. K. T. Ostovic, V. Haris, Z. Miletic, S. Lambasa, Z. Lajtman, and T. Stoos-Veic, “Fine needle aspiration cytology of metastatic merkel cell carcinoma” Collegium Antropologicum, vol. 34, no.2, pp. 691– 696, 2010. [ Links ]
11. H. Zhang, G. Gupta, X. Y. Yang et al., “A unique case of merkel cell carcinoma and chronic lymphocytic
12. Y. Yamashita, K. Toida, and H. Ogawa, “Observation of Merkel cells with scanning electron microscopy,”. Neuroscience Letters, vol. 159, no. 1-2, pp. 155–158, 1993. [ Links ]
13. M. Agelli, L. X. Clegg, J. C. Becker, and D. E. Rollison, “The etiology and epidemiology of merkel cell
14. Q. Zeng, B. P. Gomez, R. P. Viscidi et al., “Development of a DNA vaccine targetingMerkel cell polyomavirus,” Vaccine, vol.30, pp. 1322–1329, 2012. [ Links ]
15.
17. J. D. Boyer, J. A. Zitelli, D. G. Brodland, and G. D’Angelo, “Local control of primary Merkel cell carcinoma: review of 45 cases treated with Mohs micrographic surgery with and without adjuvant radiation,” Journal of the American Academy of Dermatology, vol. 47, no. 6, pp. 885–892, 2002. [ Links ]
18. B. D. Lawenda, M. G. Arnold, V. A. Tokarz et al., “Analysis of radiation therapy for the control ofMerkel cell carcinoma head and neck based on 36 cases and a literature review,” Ear, Nose andThroat Journal, vol. 87, no. 11, pp. 634–643, 2008. [ Links ]
19. P. J. Allen, W. B. Bowne, D. P. Jaques, M. F. Brennan, K. Busam, and D. G. Coit, “Merkel cell carcinoma: prognosis and treatment of patients from a single institution,” Journal ofClinical Oncology, vol. 23, no. 10, pp. 2300–2309, 2005. [ Links ]
* Especialistas en Anatomía Patológica. Hospital Dr. Calderón Guardia. Caja Costarricense del Seguro Social. Correo electrónico: pedromadriz@gmail.com
Recibido: 09 de junio de 2014 Aceptado: 30 de junio de 2014


{kind=link}
{kind=link}
{kind=link}
{kind=link}