










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina Legal de Costa Rica
On-line version ISSN 2215-5287Print version ISSN 1409-0015
Med. leg. Costa Rica vol.31 n.1 Heredia Jan./Mar. 2014
Reporte de caso
Síndrome de Noonan
Viviana Retana Gamboa1* y Laura Segura Agüero2*
Resumen:
El Síndrome de Noonan fue descrito por Noonan y Ehmke en 1963. La incidencia se ha estimado en 1 de 1000 y 1 de 2500 nacimientos vivos.1 El gen se encuentra localizado en el cromosoma 12q22 y se hereda en forma autosómica dominante y tiene una expresividad muy variable. La principal característica incluye estatura baja, defectos cardiacos, dismorfismo facial entre otros. Estatura La severidad de los síntomas varían mucho en estos pacientes. Lo que no siempre es fácil hacer el diagnóstico en los primeros años, y muchas veces son subdiagnosticados, condición que nos motivo a revisar el caso.
Palabras clave:
Síndrome de Noonan, Cardiopatías Congénitas, PTPN11: Tirosina Fosfatasa no receptor tipo 11, SHP2, Hormona de Crecimiento.
Summary:
Noonan Syndrome is a relative common autosomic dominant congenital disorder, with an incidence between 1:1,000 and 1:2,500 children worldwide. The gen is in 12q22 chromosome. The principal features include short stature, typical facial dysmorphology and congenital heart disease, among others. The range and severity of features can vary greatly in patients with NS, therefore, establishing a diagnose is difficult. The syndrome is not always identified at an early age, and many times misdiagnosed.
Keywords:
Noonan Syndrome, Congenital Heart Disease, PTPN11, SHP2, Growth Hormone.
Introducción
Asiste a la consulta de crecimiento y desarrollo un paciente masculino de dos meses de edad, vecino de Abangares. Es un niño de término, de una madre de 28 años G3P3A0 producto de parto vaginal único con un recién nacido de
Nuestro paciente actualmente tiene un año y 8 meses, no ha vuelto a presentar infecciones del tracto urinario, su leucocitosis cedió y su plaquetopenia ha mejorado, en su último control presentaba un valor de 157000 plaquetas. Un peso de 8500g y una talla de 71cm. Se mantiene en controles tanto en el EBAIS como en las consultas de cardiología, genética, endocrinología del Hospital Nacional de Niños.
Discusión
El Sindrome de Noonan (SN) es una entidad relativamente común pero con un gran subdiagnóstico, condición que nos motivó a revisar el caso.
El gen se encuentra localizado en el cromosoma 12q22 y se hereda en forma autosómica dominante, afectando a hombres como mujeres por igual, con una expresividad muy variable. En la actualidad se estima que el 50% de los SN son mutaciones de novo. 1,2.Lo que sugiere una alta tasa de mutaciones en esta secuencia específicamente en el genoma humano. Presenta heterogenecidad genética. Se han descrito 4 tipos de mutaciones en estos genes PTPN11, SOS1, RAF1 y KRAS. Se cree que el 50% son debido a mutación tipo “sin sentido” en el gen, PTPN11. 1,2,5 EL PTPN11 codifica para el no-receptor de tipo proteína tirosina fosfatasa citoplasmática SHP2, lo cual significa en una “ganancia de la función” , resultando en una exacerbación de la función original, la cual es un transductor de señal intracelular . Esta proteína intervine en la vía de señalización intracelular RAS-MAPK, implicada en el control del crecimiento, diferenciación, migración y apoptosis celular. Molécula clave en la respuesta celular de los factores de crecimiento, hormonas, citoquinas y moléculas de adhesión celular, siendo también importante en la valvulogénesis semilunar. 1,3,4,5
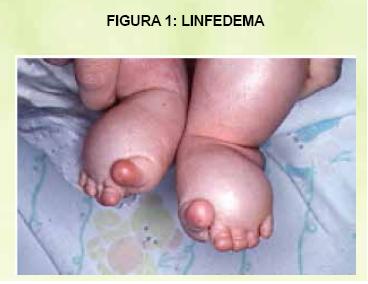
Las características faciales consisten en una cara con forma triangular, ptosis palpebral, hipertelorismo, estrabismo divergente, defectos de ambliopía, pliegue epicántico, orejas con implantación baja, puente nasal ancho y pterygium colli. Los rasgos faciales pueden irse suavizando con el edad, siendo menos evidentes en los adultos. El tórax puede presentar pectum excavatum o carinatum y escoliosis. El 68% de los pacientes presentan cardiopatías; de los cuales el 50% son de estenosis displástica de la válvula pulmonar; los defectos septales en un 20% y un 20-30% presentan cardiopatía hipertrófica, obstructiva y no obstructiva; la cual se presenta en el momento de nacer o en el período de lactante o niñez. A nivel del abdomen se observa en un 25% de los pacientes con hepatoesplenomegalia. La anomalía genitourinaria mas común es la criptorquidia en un 50% y el 50% de los varones presentan infertilidad , mientras que la función ovárica es normal, por lo que la transmisión madre-hijo es mucho más frecuente. A nivel de las articulaciones presentan hipermovilidad, cúbitus valgus y retraso en la madurez ósea de 2 años. En la piel se puede encontrar linfedema, queratosis folicular en cara y superficies extensoras, múltiples léntigos o nevos. Figura 1
A nivel neurológico encontramos hipotonía, convulsiones y retraso en el desarrollo. Los desordenes hematológicos pueden presentar deficiencia del factor IX, la enfermedad de Von Willebrand, trombocitopenia y defectos en la función plaquetaria. Los niños pueden presentar retraso del desarrollo motor en un 20%, problemas de aprendizaje en un 15%, retraso del lenguaje en un 20%, pérdida de la agudeza auditiva en un 12% además de retardo mental en hasta un 35% de los casos. 1,6. Los parámetros de crecimiento se encuentran dentro de los límites normales al nacer y la baja talla se presenta en un 80% de los pacientes entre los 2 y 4 años.10
La morbimortalidad de estos pacientes depende de la severidad de la cardiopatía congénita que presentan. En la evaluación inicial es indispensable una historia clínica con importancia al interrogatorio de enfermedades genéticas. El examen físico se realiza buscando los signos visibles. Se debe hacer un hemograma para evaluar discrasias sanguíneas. A nivel de gabinete se debe realizar un ecocardiograma, un ultrasonido de abdomen y genitales y radiografía de tórax. Es necesaria la evaluación por un genetista para determinar el diagnóstico de SN. 1 Se debe sospechar prenatalmente la posibilidad de SN en fetos con polihidramnios, derrame pleural y higroma quístico.
Clasificación de Van der Burgt es un método basado en criterios mayores y menores, para el diagnóstico del SN. 2 El diagnóstico de SN será definitivo si cumple :
Facies típicas + otro signo mayor o Facies típicas + 2 signos menores
Facies sugestivas + 2 signos mayor o Facies sugestiva + 3 signos menores. Cuadro 1
El diagnóstico diferencial es el Síndrome de Turner, Leopard, cutáneo cardio-facial, fetal por hidantoína o embriopatía por hidantoína y Neurofibromatosis Tipo I . 7,8,9 Debido a que estos síndromes cuentan con patologías cardiacas múltiples, es importante para el diagnóstico diferencial tomar en cuenta estas alteraciones.
Lo ideal es lograr dar un enfoque holístico a estos pacientes con atención multidisciplinaria del paciente. Se recomienda el asesoramiento genético si existen antecedentes familiares de este síndrome. No hay un tratamiento único para el SN. El tratamiento se centra en los problemas que se presentan. La hormona del crecimiento se ha utilizado con éxito en el SN para tratar la estatura baja. El pronóstico depende de la extensión y gravedad de los síntomas existentes. La morbimortalidad depende de la cardiopatía de fondo.
Entre algunas de las complicaciones podemos tener autoestima baja, dificultades sociales relacionadas con las anomalías físicas, infertilidad masculina en los que presentan criptorquidia bilateral, acumulación de líquido en los tejidos corporales (linfedema, higroma quístico), estatura baja y problemas de aprendizaje. Es indispensable que estos niños lleven un control estricto por un médico y en las clínicas de desarrollo.
Agradecimiento y colaboradores: Se agradece al personal del Area de Salud de Abangures por su colaboracion. Dr. Manuel Saborío Rocafort., Pediatría y Genética Médica, del Servicio de Genética Médica y Metabolismo del Centro Nacional de Prevención de Discapacidad del Hospital Nacional de Niño, por su Revisión critica.
Conflicto de interés: los autores declaran que no existe ningún conflicto de interés.
Referencias
1. J Ibrahim, Division of Genetics,
2. Jongmans M., Otten B., · Noordam K., ·
3. Takahashi I,
4. Ferreira, Souza, Arnhold, Mendonca, et al. PTPN11 mutations and response to growth hormone therapy in children with Noonan Syndrome. Journal Clin Endocrinology Metab. 2005. 5156-60. [ Links ]
5. Noordamk. Expanding the genetic spectrum of Noonan Syndrome. Horm Res 2007, paginas 24
6. Kulkarni, Ramesh. Noonan Syndrome. Indian Pediatrics 2003, 40:431-432. [ Links ]
7. Takahashi, Kogaki, Nasuno, et al. A novel mutation in the PTPN11 gene in a patient with Noonan syndrome and rapidly progressive hypertrophic cardiomyopathy. European Journal Pediatric. 2005. 497-500. [ Links ]
9. Marcy L. Schwartz; Gerald F. Cox; Angela E. Lin; Mark S. Korson; Antonio Perez-Atayde; Ronald V. Lacro; Steven E. Lipshultz. Clinical Approach to Genetic Cardiomyopathy in Children. Circulation, 1996, 2021-2038. [ Links ]
10. Rohrer t. Noonan Sydrome: Introduction and Basic Clinical Features. Horm Res 2009: paginas 3-7. [ Links ]
11. Limal J, Parfait B, Cabrol S, Bonnet D, Leceup Bruno, Lyonnet S, et al. Noonan Syndrome: relationship between genotype, growth, and growth factors. Journal of Clinical Endocrinology and Metabolism. February 2006. 300-6. [ Links ]
12. Krenz, Yutsey, Robbins. Noonan syndrome mutation Q79R in Shp2 increases proliferation of valve primordial mesenchymal cells via extracellular signal- regulated kinase ½ signaling. Circ research. 2005., 813-20. [ Links ]
13. Reardon, Donnai. Dysmorphology demystified. Arch Dis Child Fetal Neonatal Ed. 2007 May; 92(3): F225– F229. [ Links ]
14. Dr.Y Albisu. Sindrome de Turner del Genotipo al fenotipo. . Paginas 2-8. [ Links ]
15. María M. Rodríguez, Jocelyn H. Bruce, Xavier F. Jiménez, Romaguera Rita, Bancalari Eduardo, Garcia Otto, Ferrer Peter . Nonimmune Hydrops Fetalis in the Liveborn: Series of 32 Autopsies. Pediatric and Developmental Pathollogy 2005, 369-378. [ Links ]
1. Médicina General. Medicina de Empresa.
2. Medicina Física y Rehabilitación. Hospital San Vicente de Paul
1. Diseno del estudio, adquisición de la información y análisis, redacción del manuscrito.
2. Diseno del estudio, adquisición de la información y análisis, redacción del manuscrito. Trabajo realizado en Área de Salud de Abangares CCSS. Correo electrónico: vivianaretanag@gmail.com
Recibido para publicación: 16 de febrero de 2014. Aceptado: 24 de febrero de 2014


{kind=link}
{kind=link}