










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina Legal de Costa Rica
On-line version ISSN 2215-5287Print version ISSN 1409-0015
Med. leg. Costa Rica vol.27 n.1 Heredia Mar. 2010
Presentación de caso
Riñón en herradura asociado a poliquistosis renal
Dr. David Rodríguez Palomo *
Dra. Yéssica de la Paz García**
Dr. Roberto Salazar Villanea **
* Médico Cirujano, Especialista en Anatomía, Profesor del Departamento de Anatomía de la Escuela de Medicina, Universidad de Costa Rica y Universidad de Ciencias Médicas (UCIMED).
** Médico general, UCR
Resumen
Este trabajo describe la variante anatómica en un caso incidental de disección en la Escuela de medicina de la Universidad de Ciencias Médicas, el cual presenta un riñón en herradura con variante arteriovenosa que consta de cinco arterias renales y cuatro venas renales asociado poliquistosis renal.
Palabras clave
Riñón en herradura, poliquistosis renal.
Abstract
This work describes the anatomic variation from an incidental case of dissection found at Universidad de Ciencias Medicas, which presents horseshoe kidney with a right arteriovenous variant which consists of five renal arteries and four renal veins, related to Polycystic kidney.
Key words
Polyystic kidney, horseshoe kidneys.
Introducción:
Las variantes del sistema renal se presentan en un 10% de los seres humanos debido a la complejidad del desarrollo embrionario de tres sucesivas etapas, por lo cual la relación entre un riñón en herradura con la enfermedad poliquística renal presenta un incidencia muy baja, descrita desde 1/134000 hasta 1/8 millones de casos.
Dentro de las complicaciones de esta variante anatómica se encuentra la pielonefritis persistente y la hipertensión arterial refractaria por lo cual su diagnóstico se da en forma incidental, y su manejo es conservador exceptuando aquellos casos en donde la morbilidad es alta o se presente alta probabilidad de mortalidad del paciente.
Materiales y métodos:
Se utilizó un cadáver preservado de un adulto mayor de sexo femenino el cual fue disecado topográficamente. Se realiza la evisceración y disección del espacio retroperitoneal y sus estructuras. Se observa que el cadáver presentan las siguientes variantes: riñón en herradura, poliquistosis renal, cinco arterias renales y cuatro venas renales; se hacen fotografías y el estudio de las relaciones de dichas estructuras con los órganos retroperitoneales (ver Figura 1 y 2). Se realiza un análisis topográfico del hilio renal y sus componentes.
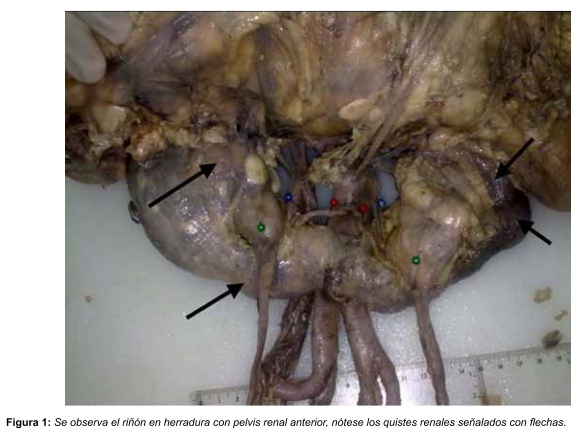
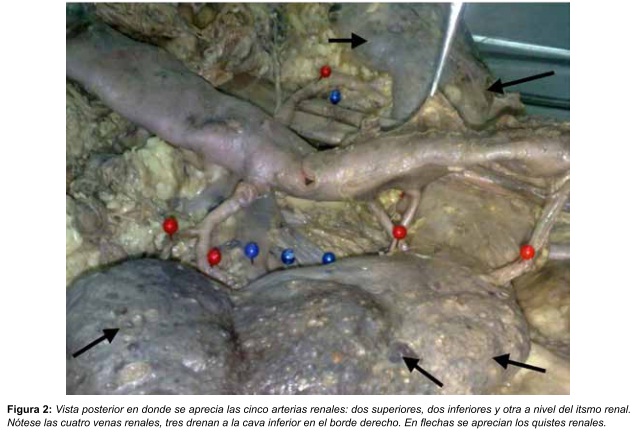
Resultados:
Se encuentra un riñón en herradura con poliquistosis renal asociado a variante arteriovenosa como describe a continuación: cinco arterias renales, dos arterias renales superiores que ingresan directo a la nivel del hilio renal, dos arterias renales inferiores que ingresan en el polo inferior del riñón y una arteria renal media que nace de la cara posterior de la arteria aorta que irriga el istmo renal (ver Figura 2).
A nivel de drenaje venoso se observan tres venas renales derechas y una vena renal izquierda, todas las venas drenan directamente a la vena cava inferior (ver Figura 2).
Discusión:
Enfermedad poliquística renal
La enfermedad poliquística renal (PKD, por sus siglas en inglés) es un desorden genético común, progresivo con afección renal primaria que se caracteriza por la presencia de múltiples quistes renales y es la cuarta causa de insuficiencia renal terminal en Australia y Nueva Zelanda. Existen dos tipos de PKD cada uno con un patrón hereditario diferente. La forma autosómica dominante afecta 1 en 400 a 1000 nacidos vivos y el tipo autosómico recesivo ocurre 1 en 10000 a 20000 nacimientos.(1,2,3) La PKD también puede encontrarse en enfermedades como el síndrome de Bardet–Biedl, la enfermedad de Von Hippel– Lindau o el síndrome de Meckel–Gruber.(4)
Existen tres procesos fisiopatológicos involucrados en la formación de quistes renales y su aumento progresivo en los pacientes con PK autosómico dominante, a) hiperplasia de células tubulares ligado a factores mediadores de la proliferación, regulación y apoptosis; b) secreción de fluido tubular disminuido debido a obstrucción tubular con enlentecimiento del flujo; anormalidades en la matriz extracelular o su función que aumenten la hiperplasia celular y la secreción de fluido.(1,4) En el 85% de los pacientes es el resultado de mutación en el gen PKD1 ubicado en el cromosoma 16 y que codifica para la proteína pilicistina 1, encargada de regular la adhesión y diferenciación de las células tubulares renales y por lo tanto, mantiene la estructura; los otros casos se dan por mutación en el gen PKD2 en el cromosoma 4, el cual codifica para la policistina 2 que es una proteína con función de canal iónico permeable para cationes incluido el calcio, cuya mutación causa la secreción de fluido dentro de los quistes. Existe un gen PKD3 que se ha relacionado con un porcentaje bajo de los casos. De forma general, las mutaciones de estas proteínas altera la función ciliar renal, que aunada a la proliferación de células tubulares causan la transformación quística.(1,2,3)
La hemorragia intraquística puede causar hematuria, sin embargo estos pacientes también tienen un alto riesgo de pielonefritis aguda, cálculos urinarios, esclerosis vascular, fibrosis intersticial e insuficiencia renal. La progresión de la PKD es causada por el proceso inflamatorio y la fibrosis secundaria en cualquiera de las formas de la enfermedad.(1)
La PKD autosómica dominante a pesar de ser la más frecuente, se presenta más tarde en la vida, diagnosticándose principalmente en pacientes de 40 a 50 años y se pueden identificar quistes progresivos uni o bilaterales. Esta forma de la enfermedad es la de menor severidad y con frecuencia no causa síntomas, por lo que el 50% son diagnosticados incidentalmente. Aproximadamente el 25-40% de los pacientes no tienen historia familiar de la enfermedad.(1,2,4) Las manifestaciones clínicas más frecuentes son la insuficiencia renal, hipertensión arterial y dolor; un 50% de pacientes desarrollan insuficiencia renal terminal a los 60 años si también presentan lesiones quísticas en hígado, vesículas seminales, páncreas y aracnoides o anormalidades no quísticas como aneurismas intracraneales, disección de aorta torácica, prolapso de válvula mitral y hernias en pared abdominal.(1,5)
La PKD del tipo autosómico recesivo es menos común pero con frecuencia es letal, con sintomatología usualmente presente desde el nacimiento o en la infancia temprana y asociada a mutaciones en el gen PKDHD1 del cromosoma 6; este gen codifica para la proteína fibrocistina/ poliductina expresada en los cilios renales y epitelio de la vía biliar. Esta forma de PKD se caracteriza por no presentar obstrucción, ser bilateral y con dilatación simétrica del 10-90% de los túbulos colectores renales.(1,2,4)
Existen manifestaciones extrarenales comunes, un 30% presentan quistes hepáticos que no afectan su función pero pueden causar dolor en el cuadrante superior derecho si son de gran tamaño o presentan sobreinfección. Los pacientes tienen una incidencia elevada de quistes pancreáticos e intestinales, divertículos colónicos, hernias inguinales y abdominales. Las alteraciones valvulares cardiacas más frecuentes son el prolapso de la válvula mitral y la insuficiencia aórtica, son detectadas en 25-30% de los pacientes. Otras alteraciones reportadas son los aneurismas principalmente del territorio coronario, aneurismas cerebrales en 4-10% de los casos con un riesgo de ruptura de 65-75% en pacientes mayores de 50 años.(1,3)
El diagnóstico de la PKD se basa en la historia clínica, antecedentes familiares, examen físico e imagenológico siendo el ultrasonido y la tomografía axial computarizada (TAC) de elección. Las imágenes permiten observar los cambios quísticos extensos e incluso la presencia de desplazamiento de tejido funcional en la proximidad de los quistes. El examen general de orina permite la detección de proteinuria y hematuria micro o macroscópica, la cual se relaciona en casos de sangrado abundante con cálculos o hemorragia secundaria a la ruptura de un quiste. Los valores de nitrógeno ureico y creatinina inicialmente son normales o levemente aumentados, pero incrementan especialmente en presencia de hipertensión arterial. Las pruebas genéticas para detección de mutaciones se reservan para pacientes con PKD sin historia familiar.(1)
La γ-glutamil transpeptidasa (γ-GT) es una enzima de membrana sintetizada en el borde en cepillo de la membrana tubular proximal, así como de células de membrana en páncreas, hígado, bazo, corazón, cerebro, vesícula seminal y endotelio. Se da documentado que la elevación de la γ-GT no se encuentra en presente en la enfermedad poliquνstica renal, lo cual hipotéticamente sucede porque los quistes derivan de túbulos que se cierran rápidamente luego de la formación de la nefrona, pero mantienen su capacidad funcional. Un incremento súbito de los niveles de γ-GT, puede significar una ruptura de los quistes con drenaje al sistema venoso.(2)
La progresión a la insuficiencia renal depende de la edad de diagnóstico, sexo masculino, pacientes de etnia negra, genotipo PKD1, volumen renal elevado y rápido, hematuria macroscópica, hipertensión arterial, quistes hepáticos en mujeres e infecciones de tracto urinario a repetición en hombres. Cerca del 10% de los pacientes mueren por enfermedad cerebrovascular hemorrágica y ruptura de aneurismas.(1,3,5)
No existe un tratamiento curativo para la PKD, sino que se enfoca en mejorar la sintomatología y dar terapia de soporte. La aspiración percutánea de los quistes puede utilizarse en casos de dolor severo por hemorragia o compresión, pero el alivio no es a muy largo plazo; por lo tanto, para dolor severo y refractario se recomienda la descompresión quirúrgica ya sea abierta o laparoscópica, pero esto no disminuye la progresión de la enfermedad. Las infecciones urinarias deben tratarse tempranamente con antibioticoterapia, para que la infección no involucre los quistes; incluso se ha descrito la nefrectomía como tratamiento de síntomas severos por infecciones recurrentes. Debe llevarse un control de la presión arterial estricto para que no contribuya con la mortalidad; incluso se demostró que la reducción de la presión arterial disminuye la incidencia de falla renal a 7 años plazo. Los estadios finales de la enfermedad renal se basan en diálisis peritoneal, hemodiálisis y trasplante, con las complicaciones inherentes de cada terapia.(1,3)
Riñón en herradura
El riñón en herradura es una malformación congénita que presenta una incidencia de 1 en 400 nacidos vivos a 1 en 800 adultos diagnosticados accidentalmente,(6,7,8,9) con una mayor incidencia en hombres en 2:1 ante las mujeres y puede ser familiar.(6,7,10,11) Constituyen la anomalía renal de fusión más frecuente (alrededor de 90%).(12,13,14)
El riñón en herradura consiste en una unión de los polos inferiores en el 95% de los casos, y en menor frecuencia se da la fusión de los polos superiores (5%) de ambos riñones mediante un istmo de tejido fibroso (15%). Puede además estar compuesto de parénquima, lo cual lo hace totalmente funcional, acompañado de una rectificación de los ejes renales y malrotación de las pelvis renales que adoptan una posición lateral y anterior.(6)
En los casos de una fusión en los polos inferiores, los riñones están malrotados, por lo que los uréteres generalmente salen del riñón en su porción ventral y el eje longitudinal de los riñones convergen.(16) Este riñón con forma de U suele situarse a la altura de las vértebras L3 a L5, debido a que el ascenso es evitado posteriormente cuando el riñón fusionado alcanza la unión de la aorta y la arteria mesentérica inferior.(6,11,12,14,15)
Dos teorías han sido propuestas para explicar embriológicamente el riñón en herradura; la teoría clásica, en donde existe un mecanismo de fusión durante la organogénesis, cuando ambos polos inferiores se ponen en contacto y se fusionan a nivel del istmo en la cara anterior de los grandes vasos abdominales. Sin embargo, este mecanismo sería válido para los riñones en herradura que presenten un istmo de tejido fibroso. La segunda teoría propuesta es que el riñón en herradura es el resultado de un evento teratogénico envuelto en la migración anormal de las células nefrogénicas posteriores, las cuales forman el istmo renal. Este evento teratogénico puede ser la explicación del riesgo aumentado de anomalías congénitas renales y la presencia de neoplasias renales.(6,10,14)
Con respecto a la asociación con otras malformaciones, en la literatura se describe que el aporte arterial está dado en un 70% de los casos por varias combinaciones de vasos simples y múltiples a nivel del hilio y el istmo renal, y un 65% de éstos por una arteria renal directa para el istmo nacida en la arteria aorta abdominal, arteria iliaca común o en la arteria mesentérica inferior,(6,10) mientras que el drenaje venoso, incluyendo el istmo, ocurre usualmente por tres vasos renales que desembocan independientemente en la vena cava.(10) Con respecto al sistema urinario, se asocia a duplicación ureteral (10%), hipospadia (5%) y riñón poliquístico, y en mujeres se asocia además a úteros bicorneos o vaginas septadas (7%) y fístula rectovaginal (2%). Entre otras malformaciones asociadas en la literatura se describe hidrocefalia (3%) con o sin mielomeningocele, enfermedad gastrointestinal, como ano imperforado (2%), malrotación y divertículo de Meckel, enfermedad cardiaca congénita de variable severidad, alteraciones vasculares, como aneurismas de aorta abdominal (17,4%) y enfermedades musculoesqueléticas (11%) como espina bífida, pie equino, paladar hendido, polidactilia (Palomo). Además, se ha asociado a trastornos cromosomales tales como Síndrome de Turner (34,8%), Síndrome de Eduards y el Síndrome de Laurence-Moon-Biedl, entre otros.(6,7,12,13,15,17)
Con base en las malformaciones asociadas y posibles complicaciones, surge una clasificación de riñón en herradura, a saber: 1) La anomalía está presente pero sin cambios patológicos y sintomatología, debido a que la unión ureteropiélica es amplia; 2) Está presente sin alteración patológica, pero presenta sintomatología, principalmente dolor abdominal superior porque el istmo del riñón comprime la aorta abdominal, hay compresión vascular y nerviosa y una variedad de perturbaciones urinarias, lo cual se conoce como enfermedad del riñón en herradura o síndrome de Rovsing; 3) Presente con complicaciones. Los síntomas son por estados patológicos debido a que los cálices están posteriores, hay una estrechez ureteropiélica.(14)
Entre las principales complicaciones que se pueden encontrar asociadas al riñón en herradura, se encuentran la nefrolitiasis (21-60%), hidronefrosis (26,1%), con o sin obstrucción de la unión ureteropélvica (15-33%), infecciones urinarias a repetición, siendo estas las más frecuentes, así como reflujo vesicoureteral (24%),(6,7,9,15,18) o con riesgo de malignización con el desarrollo de tumores renales (4,3%), como son el tumor de Wilms (más frecuente, 2-8 veces más frecuente que en la población general),(12,15) carcinoma de células transicionales de la pelvis renal, carcinoma de células escamosas, somatostatinoma, tumores carcinoides, sarcoma, linfoma, oncocitoma, colesteatoma de pelvis renal, teratoma, adenocarcinoma de pelvis renal y angiomiolipoma, siendo el carcinoma de células renales el tumor más común localizado principalmente a nivel del istmo renal.(6,15)
El diagnóstico de riñón en herradura es usualmente incidental (36%), debido a que en la mayoría de los casos cursa asintomático;(11,13) sin embargo, la presencia de síntomas y signos como hematuria, disuria recurrente, poliuria, masa abdominal y/o dolor abdominal de predominio en flancos o ángulo costovertebral, debe de levantar la sospecha de la posible presencia de un riñón en herradura.(6)
Para su estudio se utiliza el ultrasonido abdominal, que demuestra la presencia del istmo o una banda de tejido renal que conecta los dos polos renales o cuando se observa que los ejes renales convergen, "riñones pequeños" o una masa preaórtica;(16) y el PIV (Pielograma Intravenoso), en el cual se puede observar una rectificación axial, malposición piélica y la presencia de los cálices del grupo inferior que convergen y se colocan mediales a los uréteres. En ocasiones se usa el TAC para evaluar el istmo y posibles masas sospechosas, o bien las exploraciones más invasivas como la ureteropielografía retrógrada que permite estudiar los puntos de obstrucción o la laparoscopía exploratoria para descartar tumores.(6)
El tratamiento quirúrgico está indicado cuando tiene lugar con nefrolitiasis recurrente con hidronefrosis secundaria, obstrucción de la unión ureteropiélica y reflujo vesicoureteral complicado. (12,14)
Enfermedad poliquística en riñón en herradura
La asociación del riñón en herradura con la enfermedad poliquística renal es bastante inusual, con rangos de incidencia que van desde 1/134000 hasta 1/8 millones de casos.(8,19,20) Las complicaciones de estas anomalías renales incluyen lumbalgia, infección y malignidad. El riesgo de desarrollar una falla renal no es mayor que cuando se presenta sólo el riñón en herradura. (19) Hasta la fecha, no se ha descrito ninguna asociación genética entre las dos patologías.(20)
El diagnóstico se puede realizar principalmente con un ultrasonido abdominal y TAC, así como el PIV.(19) El manejo terapéutico de estos casos está basado eminentemente en el control de las manifestaciones clínicas propias de la insuficiencia renal que presentan estos pacientes.(8) El tratamiento quirúrgico, el cual incluye la nefrectomía bilateral, se encuentra indicado cuando el paciente presenta pielonefritis persistentes, hipertensión refractaria y en preparación para el trasplante renal.(19)
Recibido para publicación: 12 de enero de 2010 Aceptado: 27 de febrero de 2010
Bibliografía:
1. Stawicki, S y Lombardo, G. Polycystic kidney disease. Scientist. 2008; 2(1):17-20. [ Links ]
2. Taşçı, İ; Bulucu, F; Mas, MR; Verim, S. Polycystic kidney disease with highly elevated γ-glutamyl transpeptidase. Eur J Gen Med. 2005; 2(1):39-40. [ Links ]
3. Thomas, MC. Autosomal-dominant polycystic kidney disease. Nephrology. 2007; 12: S52–S56. [ Links ]
4. Simons, M y Walz, G. Polycystic kidney disease: Cell division without a c(l)ue?. Kidney International. 2006; 70: 854–864. [ Links ]
5. Ferrero, R; Moreno, F; Calatrava, S; García, F; Gassó, M; Díaz, E. Enfermedad poliquística del adulto en riñón en herradura. Actas Urol Esp. 2004; 28(3): 243-244. [ Links ]
6. Rodríguez, D. Riñón en Herradura Asociado a Variantes Anatómicas, Medicina Legal de Costa Rica.2009;26(1):73-80. [ Links ]
7. Weiser, AZ., et al. Determining the Incidence of Horseshoe Kidney from Radiographic Data at a Single Institution. The Journal of Urology.2003;170:1722-1726. [ Links ]
8. Ferrero, R., et al. Enfermedad Poliquística del Adulto en Riñón en Herradura. Actas Urol Esp.2004;28(3):243-244. [ Links ]
9. Kao, PF., et al, The 99mTC-DMSA Renal Scan and 99mTC-DTPA Diuretic Renogram in Children and Adolescents with Incidental Diagnosis of Horseshoe Kidney. Nuclear Medicine Communications, 2003;24:525-530. [ Links ]
10. Tijerina de la Garza, O., et al. Anatomical Study of the Horseshoe Kidney Int. J. Morphol.2009;27(2):491-494. [ Links ]
11. Martinez-Mier, G., Rayhill, S., Katz, D., El Trasplante Cadavérico de Riñón en Herradura: Reporte de Dos Casos, Cir Ciruj.2005;73:211-215. [ Links ]
12. Romero, FJ., et al, Anomalías Renales de Número, Posición, Forma y Orientación: nuestra experiencia. Vox Paediatrica.2003;11(2):16-26. [ Links ]
13. Ferreira, HH., León, J., Patología Infantil en el Paciente Adulto Rev Colomb Radiol.2008;19(4):2522-2527. [ Links ]
14. Lozano, R., Riñón en Herradura, Presentación de un Caso y Revisión. Rev Med Hond.2000;68:105-109. [ Links ]
15. Yohannes, P., Smith, AD., The Endourological Management of Complications Associated with Horseshoe Kidney. The Journal of Urology.2002;168:5-8. [ Links ]
16. Hahm, AM., Ritz, E., Renal Ultrasonography Lesson: Horseshoe Kidney. Nephrol Dial Transplant.1999;14:2740-2741. [ Links ]
17. Garza, ME., et al, Riñón en Herradura con Doble Sistema Colector Completo Derecho, Reporte de un caso. Rev Mex Urol.2004;64(5):239-241. [ Links ]
18. Cascio, S., et al. Vesicoureteral Reflux and Ureteropelvic Junction Obstruction in Children with Horseshoe Kidney: treatment and outcome. The Journal of Urology.2002;167:2566-2568. [ Links ]
19. Shahreyar, M., et al. Polycystic horseshoe kidney. Indian J Nephrol 2005;15: 250- 251. [ Links ]
20. Batista, LA., et al. Polycystic horseshoe kidney. Nephrol Dial Transplant (2007) 22: 652–653. [ Links ]

