











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introduction
Stem cells (SC) are the main focus in a lot of research for the development of novel treatments for tissue regeneration. These cells are able to: migrate to damaged tissue and participate in wound healing process; produce many growth factors (GF) that induce and modulate regeneration mechanisms; differentiate into various cell types and immunomodulate. Altogether, playing a fundamental role for generation of de novo tissue and avoid scars (1) (2) (3).
Given the significance of stem cells, it is required manipulate them for expansion, cryopreservation and also to analyze biochemical, genetic, molecular, safety and efficacy properties to determine potential therapeutic applications (1). Promising results have been obtained through research in regeneration and autoimmune diseases (4), further characterization, analysis of their behavior and ability to differentiate in vitro are mandatory prior to move forward with preclinical and clinical research (5).
Adipose tissue is composed mainly of mature adipocytes (90%), but it is possible to identify a heterogeneous population of cells, called Vascular Stromal Fraction (SVF) which includes: preadipocytes, progenitor endothelial cells, pericytes, mast cells, macrophages, fibroblasts and around 2% are adipose tissue stem cells (ASC) that can differentiate into several lineages (6) (7) (8) (9). ASC are similar to mesenchymal stem cells from bone marrow, which act as precursor cells and help to maintain the tissue around them (10). Many reports support the ability of ASC to differentiate into cells of the mesodermic lineage (bone, fat, cartilage and muscle) at in vitro level controlled by hormonal, neuronal and paracrine factors (11). However, transdifferentiation of ASC to ectodermal or endodermal lineages has also been demonstrated through several markers (12) (13) (14) (15).
ASCs can be isolated by minimally invasive procedures and the yield of ASCs obtained from adipose tissue is 10% from a pool of 1 million of nucleated cells isolated (whereas 1% is recovered from a sample of 1 million nucleated cells from bone marrow or less if isolated from peripheral blood) (16) (17). Additionally, they can be prepared under Good Manufacturing Practices (GMPs) standards, and can be used for cellular therapies even without being cultured and expanded (applying the SVF) which reduces the risk of excessive handling or potential contamination and costs (13) (18) (19). These cells do not express HLA-DR (16), so they can be used in allogenic manner, they inhibit the proliferation of peripheral blood nucleated cells and decreases the immunological response (11) (20). On the other hand, in vivo studies have shown that ASC or SVF applications in skin lesions promote and increases the regeneration rate, decrease pain, inflammation, and the obtained tissue is more comparable to normal skin than scar formation (21) (22) (23) (24). Currently, there are a considerable number of clinical studies in different stages applying these cells (25) (3) (26).
Before considering them as candidates for therapies, the International Society for Cellular and Gene Therapy (ISCT) proposed the following criteria: adherence to plastic, more than 95% of the population must be CD73, CD90 and CD105 positive, and negative for CD34, CD45, CD11b or CD14, CD79 or CD19 and HLA class II. Additionally, they must be able differentiate in chondrocytes, osteoblasts and adipocytes (5). The aim of this study was to isolate and expand ASC from adipose tissue that meet the ISCT stem cells criteria for cell therapy.
Material and methods
Assessment of ASC derived from murine adipose tissue in in vitro culture
Culture conditions
All animals were handled under current legislation and permits established by the Institutional Committee for the Care and Use of Animals (CICUA) of the UCR (CICUA-047-16). Balb-C mice were supplied by the Biological Testing Laboratory of the University of Costa Rica (UCR) at 4 weeks old. Adipose tissue from inguinal area was isolated from each mouse after euthanasia was performed using cervical dislocation during anesthesia. The in vitro culture experiments were carried out at Center for Biotechnology Research at Costa Rica Institute of Technology (TEC), Cartago. The samples were collected in PBS and washed with PBS + 5% Penicillin and Streptomycin (GIBCO). Mechanical and enzymatic disaggregation were performed with 0.1% type I collagenase (GIBCO) solution incubating the samples at 35 °C for 60 minutes. The suspension was inactivated with Fetal Bovine Serum (GIBCO) and centrifuged for 6 minutes at 1200 rpm. The pellet was resuspended in Dulbecco’s modified Eagle’s Medium (DMEM) high in glucose (4g/L) (GIBCO) with Ham’s F12 (SIGMA) 1:1 supplemented with + 10% Fetal Bovine Serum (GIBCO), 1% L -glutamine (GIBCO) and 1% antibiotics (GIBCO). The cells were incubated at 37 °C with 5% CO2 and medium was renewed every 3-4 days. Subcultures were performed when the confluence reached 80%.
Cryopreservation
Two different cryopreservation methods were evaluated a mix of 10% dimethylsulfoxide (DMSO) + 30% SFB + 60% culture medium and a mix of 5% DMSO + 30% SFB + 65% culture medium. The thawing process was also evaluated by two different methods: in the first one, the vial was heated in a water bath at 37° C, placed directly in the culture flask with fresh medium and incubated under normal conditions. The second method consisted of slightly thawing the vial in a water bath at 37ºC and diluted in 9mL of fresh media, then centrifuged for 6 min at 1200 rpm and the resulting cell pellet was resuspended in fresh medium and placed in a culture flask under normal incubation conditions.
Cell kinetics
Three different cell growth curves were made using passages 1, 2 and 3. Initially 4x103 cells were seeded per well on a 12-well plate and totsl cells were counted every two days for 12 days. Each assay was performed in triplicate. Additionally, the population doubling time (PDT) was calculated. For this, 1x105 cells were seeded in a T25 flask and allowed to 90% confluence and the next formula was applied.
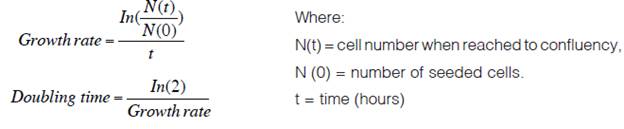
Identification of ASC markers
Four markers were selected to identify the ASC population from passage 1 by flow cytometry: anti-CD45 PE, anti-CD73 PerCP, anti-CD90 FITC, anti-CD105 Alexa 647 (BD Biosciences). A cell suspension was made, and the cells were incubated with the antibodies at an established concentration for 20 minutes in darkness. Afterwards, the sample was washed with FACS buffer and analyzed with the BD Accuri C6 flow cytometer.
Differentiation potential of the ASC
P3 cells were cultured in a 24 multiwell plate using the Mouse Mesenchymal Stem Cell Functional Identification Kit (R&D Systems, #SC010), according to manufacturer’s instructions. These cells were maintained in culture for 21 days with the three-different induction’s media, renewing them every 3-4 days.
Immunofluorescence was performed on the differentiated cells as follows: cells were fixed in 4% paraformaldehyde for 20 min, washed with a blocking solution and subsequently incubated with anti-FABP4, anti-Collagen type II or anti-Osteopontin overnight at 2-3ºC, to demonstrate the differentiation to the adipogenic, chondrogenic or osteogenic lineage respectively. Then, they were incubated with secondary antibody in the dark. Finally, Prolong Antifade mounting medium with Dapi (Thermofisher) was added and they were analyzed on the Olympus CKX41 fluorescence microscope.
Results
Assessment of ASC in vitro culture from murine adipose tissue
A total of 11 effective isolations were carried out, obtaining 0.8 ± 0.2 g of adipose tissue per sample. After several modifications and adjustments to the protocol, 1.7x105 ± 4x104 nucleated cells were counted from each processed tissue. When seeded, they first formed microcolonie, with a spindle-shaped morphology, and reached confluence after 10-14 days in culture (Figure 1A). When the cells reached 70% -80% confluence, they were subcultured on a 1:2 - 1:3 ratio (Figure 1). The maximum passage reached was P5, then they began to show signs of stress: they changed from a fibroblast-like morphology to a starry morphology, vacuoles could be observed inside the cytoplasm and their confluence couldn’t surpass 40% (Figure 1E).
Regarding the freezing/thawing evaluated methods, the most efficient medium for cryopreservation was 5% DMSO, 30% FBS and 70% DMEM/F12 culture medium. On the other hand, cells cryopreserved with 10% DMSO were not able to adhere to the culture plate afterwards.
The most successful defrosting method was the one that eliminated the DMSO by centrifugation after thawing. Cells thawed under these conditions, regrow in vitro with the same efficiency and kinetics (Figure 2) as no cryopreserved cells, while those that were thawed by direct seeding did not proliferate when seeded (images not shown).
To analyze the behavior of these cells in vitro, cell kinetics tests were performed. A growth curve was made in passages 1, 2 and 3 as the PDT analyzed (Figure 3).
Very similar lag phases were obtained among all the passages. As the number of passages gets higher, there is a slight increase in the exponential phase. Finally, the stationary phase was reached near 2.4x105 ± 1x104 cells/well or 6x104 cells/cm2 in all cases. The population doubling time range was maintained between 35.68 - 39.82 hours with the established incubation conditions (Figure 6). The ANOVA test showed significant differences between P1 and P2 (p = 0.02), as well as P1 and P3 (p = 0.04).
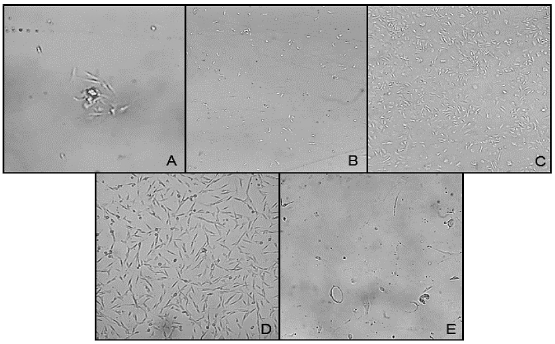
Figure 1 Stromal cells derived from adipose tissue in culture at different timepoints. A. Day 1 (100X), B. Day 5 (40X), C. Day 10 (40X), D. P2 cells (40x), E P5 cells (100X). Abbreviation: P, passage.
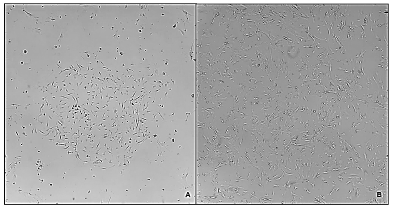
Figure 2 Morphology of P3 stromal cells derived from adipose tissue after cryopreservation and thawing process. A. Day 1 after thawing, B. Day 4 after thawing. (40x).
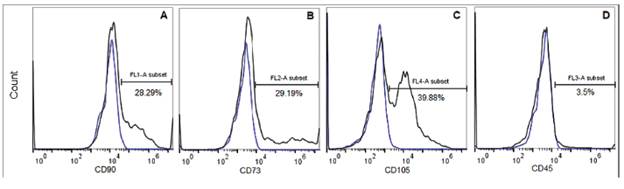
Identification of ASC markers. A passage 1 population was analyzed for stem cell markers expressions. As shown in figure 4, it can be seeing that, close to 30% of the cells, express CD73 and CD90 (Figure 4.a and 4.b), and approximately 40% of analyzed population was CD105 positive (Figure 4.c). Whereas, the 3,5% of the population express the CD45, which means that only these cells come from hematopoietic linage (Figure 4.d).
Differentiation potential of the ASC.Figure 5 shows the immunofluorescence of the cells after 21 days of culture in the respective differentiation media. For adipogenic, differentiation the fatty acid binding protein-4 (FABP4), commonly used for adipose marker is shown; for the chondrogenic differentiation type II Collagen (Col II) was used and finally for osteogenic induction, the Osteopontin (OPN), a bone specific protein synthetized by osteoblast was detected (27). Besides the immunostaining of different markers, a morphological change could be noted. The cells committed to adipogenic linage, increase their size up to 3 times, showing vacuoles within the cytoplasm. On the other hand, the cells that were subjected to chondrogenic differentiation, showed a starry morphology, did not increase their size as much as the cells in adipogenic induction, and type II collagen was synthesized and accumulated perinuclearly. In parallel, the cells that were directed to osteogenic differentiation retained a more fibroblastoid morphology, smaller size compared to the last two and osteopontin could be located at the nuclear perimeter.
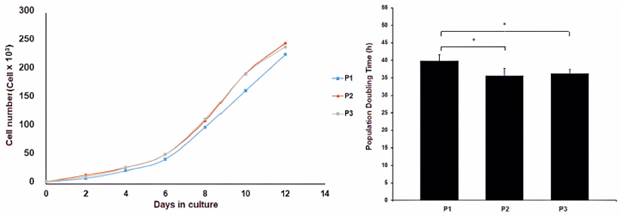
Figure 3 A. Growth curves of the ASCs cultured in vitro at different passages, B. Population doubling time of ASCs at different passages
Discussion
Due to the great regenerative potential of the ASC for wound healing, a protocol for their isolation from a minimal sample of adipose tissue was proposed. In this paper, we present a feasible protocol that allows a good isolation yield of ASC from a small sample, thus demonstrating that adipose tissue is an excellent source of mesenchymal stem cells. To the date, there is no gold standard method for the isolation of these cells. In addition to the protocol variations and the reagents used, there are many variables that can affect the amount and composition of isolated cells from fat deposits such as the method of obtaining adipose tissue, age and sex of the donor, body mass index, anatomical area from which the sample is extracted, physiological status of the donor among others (28).
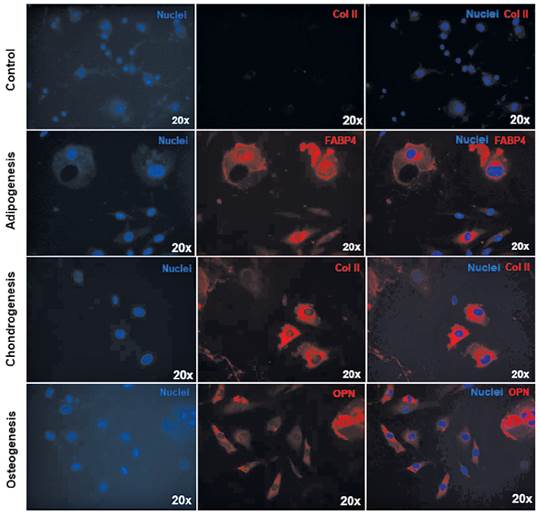
Figure 5 Analysis the multipotential of the ASCs. Differentiation in adipogenic lineage (FABP4 in red), chondrogenic (type II collagen in red) and osteogenic (osteopontin in red). Nucleus of cells in blue. The control was performed for each case, however only the respective chondrogenic differentiation is shown here.
The number of isolated cells varied according to the volume of the obtained sample. In this case, it was obtained 1.7x105 ± 4 x 104 nucleated cells per each 0.8 g of fat tissue. Bear and colleagues (15), have reported higher amounts of isolated cells using human adipose tissue, 0.5-2.0x106 cells per gram. However, in a study conducted by Bobis and colleagues, they demonstrated similar amounts to those reported in this study when using collagenase, but when using another enzyme, Liberase ™ (Roche), the performance was higher (29). Thus, the used disaggregation enzyme could improve or diminish the yield per volume from the same sample. The number of isolated cells varies also regarding the anatomical zone from which the subcutaneous adipose tissue was taken, the type of adipose tissue (subcutaneous, visceral or brown), and the physiological condition of the donor. This was slightly observed in preliminary tests with older mice, where even though they had more adipose tissue, the number of isolated cells did not reach 100 000 cells (data not shown).
Once the cells were isolated and cultured in vitro, they were able to maintain up to 5 passages, which results consistent with a study carried out by Fajka-Boja and colleagues in 2018 (30). A study made with samples obtained from rabbit reached a maximum of 7 passages (31) however, there are multiple studies in which the culture medium was supplement with growth factors reaching up to 14 passages and more. It is necessary to consider that due to the limited size of the initial sample the cells seeded for this study were less, which means that they performed more doublings than in other studies where the seeding density was higher. Therefore, although there are few passages in this case, using the PDT as an indicator to compare to experimental results is more appropriate (32). The PDT reported in literature for ASC varies from 20 to 120 hours and is influenced by many variables (33) (34). Hwa and colleagues found that at low passages, they ranged between 1.6 and 1.7 days and as the passages increase, this value does the same (35). On the other hand, Blázquez and colleagues reported a range between 6.4 - 14.7 days using a commercial medium specific for ASC (36) and when using mesenchymal stem cells from umbilical cord, they presented the shortest PDT, indicating that beside all the mentioned factors, the kind of tissue from which the mesenchymal stem cells are obtained affects the kinetics. Beane and collaborators also studied the age of the donors and found that the PDT of the ASC was affected by it, finding samples of different ages with values of 20 hours (34).
During this test, cells that underwent a cryopreservation process using 10% DMSO did not survive. However, by reducing it to 5% in the cryopreservation media and eliminating it when the cells were re-cultivated (second thawing method), the mortality was reduced considerably. That confirms the toxicity of this cryoprotective agent to ASCs, being a toxic substance at room temperature that damages cells when used at high concentrations or inadequately. Other researchers have reported that cryopreservating cells using DMSO at 10 % concentration within the medium has caused adverse effects in patients that receive stem cells as therapy, such as: respiratory reactions and neurotoxicity in pediatric patients, which clearly indicates the need to reduce its use or look for other cryoprotective agents when aiming to scale up to clinical therapies (37) (38).
The ISCT defined that cells in vitro must be positive for CD73, CD90 and CD105, but negative for CD45 and HLA-DR markers as identifiers for mesenchymal stem cells isolated from various tissues (fat, bone marrow, umbilical cord, blood and other). Several authors, however indicate that it is not necessarily a manifestation of them in vivo (32). In this study, only around 25% of the analyzed cells showed to be positive for the CD73, CD90 and CD105 markers, while 96% were negative for the CD45, consistent with the criteria from ISCT. This analysis was assessed with low passage cells, therefore it can be noticed that in this stage, the cell culture is not homogeneous yet, but it contains the population of interest, and probably, as the passages and doublings increase, the percentage of this population will too, and also the homogeneity of the cells. The reason to analyze a low passage cell sample was that after multiple subcultures the manipulation increases, which increase the risk of contamination, change their phenotype and the cells can accumulate cariotypic abnormalities after the constant cell divisions (39). Despite this, it is desirable to analyze each of the passages to determine at what specific stage it is possible to obtain at least one population greater than 90% that presents the conditions indicated by the ISCT. Other studies performed with rat cells at passage 4, showed that 96% were positive for CD90 and negative for CD45 (40). In addition, a study comparing different methods of ASC isolation showed that the expression profile varies according to each protocol. In some cases, they only showed expression of CD90 and CD105, while in other cases only the expression of CD90 and CD73 was found, but always were negative (<5%) for CD45 (41).
This could be cause by using different tissue sources, which favors the isolation of different subpopulations of mesenchymal stem cells, or that the process of isolation and cultivation affect the expression of them. Although there are multiple investigations that found the expression of all the markers contemplated by the ISCT (13) (42), it is possible to find different reports (39) (43).
A fundamental characteristic of the stem cell is its capacity to divide asymmetrically, where one descendant cell will be identical to the cell of origin and the other will acquire characteristics of a specialized cell (39). This differentiation will occur when the adequate stimuli trigger the process. In this study, differentiation of isolated cells could be accomplished into adipogenic, chondrogenic and osteogenic lineages. These results were consistent with other studies developed that assure their differentiation potential (44) (45) (46). Meeting the stem-cell definition criteria established by ISCT (33).
Conclusions
The established protocol was effective for isolation of ASCs, and culture could be maintained in vitro until 5 passages, afterwards the cells entered in a senescence state. Cellular kinetics matched the results obtained in literature. The best cryopreserving method was the one with the lowest concentration of DMSO, and the best thawing method was eliminating the DMSO before seeding. A third of the P1 population cells analyzed by flow cytometry showed stem cell markers. However, it is necessary to subculture the cells repeatedly in order to obtain more cells with these characteristics and a more homogeneous culture.
P3 cells were able to differentiate into three lineages: adipogenic, chondrogenic and osteogenic, as shown by the presence of tissue-specific proteins and morphological changes, which meet the ISCT criteria to determine the identity of an adipose-derived mesenchymal stem cells culture.

