











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La Bioinformática ha demostrado obtener mejoras en la investigación en áreas afines. Biólogos usan programas para alinear secuencias y crear estructuras, que son de importancia para analizar los resultados dados por dichos programas de software. De esta manera, se ha logrado descubrir especies nuevas, determinar a qué especie pertenece una muestra de un ser vivo, curas, entre otros descubrimientos.
Determinar a qué especie pertenece un ser vivo, o descubrir una nueva especie, no es trabajo fácil; no es solo que la muestra en análisis y la especie conocida coincidan morfológicamente, sino que también, sus características genéticas se relacionen de alguna forma para poder demostrar que una muestra pertenece a una especie existente. Para identificar una muestra, debe hacerse una extracción de ADN, y una vez obtenidas sus secuencias, efectuar el análisis con ayuda de programas como bioEdit, genDoc, Mega, Autodimer, Dnasp5 (para alinear y analizar secuencias de ADN), PAUP, Mr. Bayes, Beast y Dambe (para la creación de árboles filogenéticos). Luego, los resultados obtenidos se analizan.
En este artículo se trabajó con un grupo de muestras de una especie desconocida. Según el estudio morfológico realizado a dicha muestra, pertenecía a la especie lepidópteros; sin embargo, esta relación no se podía aceptar como un hecho pues un estudio de este tipo puede no dar resultados ciertos.
Alineamiento de secuencias de ADN
Según el modelo propuesto por Watson y Crick, la molécula de ADN está compuesta por dos hebras helicoidales (Torres, 2007), cada una siguiendo una secuencia de nucleótidos {A;T;C;G}, y formando parejas de {C;G} y {A;T} entre las dos hélices (Ferrández, Hernández & Pastor, 2003).
Una vez extraído ADN de una muestra, se obtienen dos secuencias de ADN (forward y reverse). Deben guardarse cada una en un documento con formato FASTA (Figura 1). Se debe realizar un BLAST a la secuencia forward, que consiste en hacer una búsqueda en una base de datos de secuencias de ADN, para encontrar datos similares y verificar si el amplificador es correcto (Altschul, Madden, Schäffer1, Zhang, Zhang2, Miller2 & Lipman, 1997).
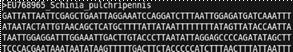
Bioedit
Bioedit es un editor de alineamiento de secuencias, sumamente amigable con el usuario (Hall, 1999). Con este programa, se realizaron tres pasos importantes: reverso complementario, algoritmo Smith-Waterman y algoritmo Needleman-Wunsch.
Reverso complementario: El reverso de una secuencia simplemente consiste en revertir la hilera dada, y el complemento, en cambiar los nucleótidos A con T o C con G, y viceversa (Figura 2).
El reverso complementario convierte una secuencia de ADN en su reversa y luego aplica el complemento. Se realiza a cada secuencia reverse de las muestras de ADN. Los pasos para realizar esto en el programa son los siguientes: Sequence->Nucleic Acid->Reverse Complement.

Algoritmo Smith-Waterman: Este algoritmo de alineamiento de secuencias local encuentra y alinea las regiones similares entre dos hileras (Smith y Waterman, 1970), con el fin de asegurarse de que la secuencia de ADN realmente es la óptima, y coinciden un cien por ciento.
En este paso, aplicados los cambios anteriores a las secuencias reverse, se realiza el alineamiento con sus secuencias forward correspondientes. Las instrucciones para ejecutar el algoritmo son las siguientes: Alignment ->Local Alignment.
Algoritmo de Needleman-Wunsch: El algoritmo de alineamiento global, se basa en alinear y mostrar dónde calzan en forma perfecta o casi perfecta las secuencias (Needleman y Wunsch, 1970).
Una vez ya alineada cada muestra con su forward y reverse, se ejecuta en todas las secuencias editadas. Las instrucciones para ejecutar el algoritmo son las siguientes: Accessory Application->ClustalW Multiple Alignment. Finalmente, se deben guardar todas las secuencias juntas y alineadas en un documento formato FASTA.
Es importante realizar los pasos anteriores, debido a que los resultados obtenidos servirán para un análisis con menor riesgo de errores, y mayor precisión. Mediante la aplicación de los pasos anteriores en el análisis de las secuencias de ADN de lepidópteros, se demostró que todas pertenecían a una misma especie, ya que presentaban pocas mutaciones y eran muy similares.
Árboles filogenéticos
La filogenia molecular y aplicaciones como PAUP, Mr. Bayes y Beast, entre otras, son usadas en investigaciones comparativas en genética. Algunos estimadores filogenéticos se basan en un modelo explícito de la evolución de nucleótidos para estimar parámetros evolutivos tales como longitudes de rama y topología del árbol. Los árboles filogenéticos son generados para analizar las relaciones evolutivas observadas y obtener información a partir de ellas, de manera que facilitan encontrar la divergencia de linajes, o la relación entre ellos (Bos y Posada, 2005).
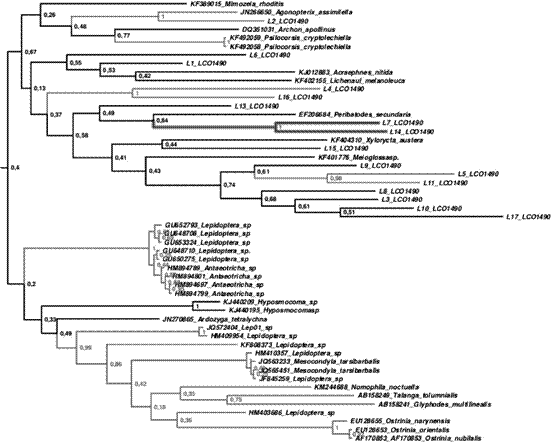
Para la creación del árbol filogenético de las muestras de ADN obtenidas, morfológicamente parecidas a lepidópteros, con el fin de determinar si había relación con las secuencias de ADN de lepidópteros, obtenidas de una base de datos, se utilizaron los programas PAUP, Mr. Bayes y Beast.
Se obtuvo un árbol filogenético (Figura 3) que demostró que las muestras no pertenecían a la especie de los lepidópteros, debido a que no presentaban relación alguna con esta especie, pero sí gran similitud entre ellas. Al parecer, se reveló el descubrimiento de una nueva especie, actualmente con nombre desconocido.
Por otro lado, con el uso de estos programas de software, se concluyó que no son tan amigables con el usuario que no tiene conocimiento de comandos de línea.
Conclusiones y recomendaciones
Se recomienda para analizar grandes cantidades de datos utilizar aplicaciones que procesan y ejecutan algoritmos eficientes, para generar resultados con mayor precisión y menor riesgo de errores.
Es importante mejorar las aplicaciones de Bioinformática que han sido producidas por expertos en campos diferentes a la Ciencia de la Computación, ya que muchas veces se desaprovechan las posibilidades de optimización (e.g., uso máximo del hardware disponible, metodologías de desarrollo de software apropiadas, lenguajes más eficientes, etc.).

