










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Ciencias Médicas
Print version ISSN 0253-2948
Rev. costarric. cienc. méd vol.19 n.3-4 San José Dec. 1998
Resumen
Los informes de la evaluación cinética de los medicamentos desarrollados en los últimos 20 años documentan, prioritariamente, los resultados de ensayos realizados exclusivamente con varones; o bien, con muestras mixtas pero cuyos resultados no se reagrupan ni analizan por la variable sexo. El objetivo de este ensayo fue evaluar la influencia del sexo en el comportamiento farmacocinético de una benzodiacepina (loracepam) en sujetos sanos. Se administró una dosis oral de 2 mg de loracepam (LOR) a 7 varones (V) y 8 mujeres (M) sanos; se extrajeron muestras sanguíneas antes y después de la administración del fármaco a diferentes tiempos; se cuantificaron las concentraciones plasmáticas (Cp) con HPLC y para cada sujeto se calculó sus parámetros cinéticos. Los resultados mostraron que la concentración máxima (CMAX) fue de ± 19.66 ng/mL (V= 16.28 ± 2.51 ng/mL, M= 23.04 ± 6.12 ng/mL), los tiempos para alcanzar CMAX (tMAX) oscilaron entre 1h (n= 7, V=3, M= 4) y 1,5 hs (n= 4, V= 2, M= 2). El tiempo de vida media (t1/2) de eliminación fue de 11.33 hs (V= 11.98 ± 2.39 hs, M= 10.68 ± 2.69 hs) y el Area Bajo la Curva de 0 a 48 hs (ABC0-48) alcanzó una magnitud de 199,46 ng/mL·h (V= 168.36 ± 28.09 ng/mL·h, M= 230.57 ± 100.38 ng/mL·h). A partir de +1h las Cp resultaron más elevadas en mujeres, siendo significativamente mayor a +1.5hs (V= 14.69 ng/mL, M= 19.18 ng/mL; p= 0.033) pero los diferentes parámetros evaluados no se diferenciaron en función del sexo. Todos los sujetos mostraron una buena tolerabilidad al fármaco. En conclusión, se registra una tendencia a la diferenciación en la farmacocinética del LOR atribuible al factor sexo, pero es necesaria la realización de ensayos con muestras de mayor tamaño para establecer una diferenciación real y significativa. Es procedente realizar análisis cinéticos con los diversos medicamentos para explorar y describir posibles diferencias cinéticas atribuibles al sexo y analizar su repercusión clínica.
Palabras clave
loracepam, farmacocinética, diferencias por sexo.
Abstract
Pharmacokinetic evaluation of new drugs during the last 20 years presents results of clinical trials performed with males or both sex samples without sex-related description and analysis.
The aim of this trial was to investigate the sex-related pharmacokinetic differences of benzodiazepina (lorazepam) in healthy subjects of both sexes.
A single oral dose of lorazepam 2 mg was administred 7 males (M) and 8 females (F). Blood samples to assess lorazepam plasma concentration by HPLC were obtained before and at different times following drug intake. Kinetic parameters were calculated for each subject.
Peak levels (CMAX) were 19,66 ng/mL (M= 16,28 ± 2,51 ng/mL, F= 23.04 ± 6.12 ng/mL). The time to reach maximum concentration (tMAX) was between 1h (n= 7; M= 3, F= 4) and 1,5 hs (n= 4; M= 2, F=2). The biological elimination half-life (t1/2) was 11,33 hs (M= 11.98 ± 2.39 hs, F= 10.68 ± 2,69 hs) and the Area Under the concentration-time Curve 0 to 48 hs (AUC0-48) reached a maximum of 199,46 ng/mL·h (M= 168,36 ± 28,09 ng/mL·h, F= 230,57 ± 100,38 ng/mL·h). At +1h females showed higher values than males, significant differences in lorazepam levels were observed only at +1.5hs (M= 14,69 ng/mL, F= 19,18 ng/mL, p= 0,033) but no significant differences was obtained when comparing pharmacokinetic parameters by sex. Lorazepam was well tolerated by all subjects.
In conclusions, the analysis of pharmacokinetic parameters by sex with lorazepam showed as light variation, new trials with larger samples must be made to determine whether or not the pharmacokinetic profile is sex-dependent. The perfomance of clinical trials to evaluate sex-related differences is very important to help optimize clinical use and drug prescription.
Key words
lorazepam, pharmacokinetics, sex-related differences, healthy subjects.
Introducción
Los estudios farmacocinéticos se realizan fundamentalmente durante la Fase I de Investigación en el curso del desarrollo clínico de un fármaco. Primordialmente por seguridad, sobre todo cuando es la primera administración en humanos, estos ensayos clínicos suelen incluir sujetos masculinos, de modo que los análisis se basan en resultados obtenidos de una muestra que representaría solo una fracción de la población. Por su parte, los estudios en fase de Investigación siguientes (Fase II y III) incluyen muestras con pacientes de uno y otro sexo, pero el énfasis respecto a la eficacia que tienen tales ensayos no suele acompañarse del análisis cinético de los tratamientos objeto de estudio (1-3).
En general, la evaluación cinética documentada en la literatura referente a los fármacos desarrollados en los últimos 20 años informa, prioritariamente, resultados de estudios realizados exclusivamente por varones; o bien, con muestras mixtas pero cuyos resultados no se reagrupan ni analizan por la variable sexo (1-4), lo que representa una limitante real al momento de la extrapolación y generalización de resultados a la población general. Diferencias farmacocinéticas, farmacodinámicas y en la incidencia de efectos adversos debidos a drogas han sido informadas para una gran cantidad de medicamentos (4), lo cual brinda pertinencia a los estudios diseñados para evaluar este aspecto.
En el caso particular de los psicotrópicos, la información disponible tiende a enfatizar el análisis de los aspectos dinámicos, debido a las implicaciones que tienen sus efectos sobre el sistema nervioso central (3,4). Por tanto, el presente trabajo se llevó a cabo para describir el perfil cinético tras la administración de una dosis única de loracepam (LOR) y evaluar posibles diferencias cinéticas debidas al factor sexo.
Materiales y métodos
El protocolo y los procedimientos experimentales se ajustaron a las directrices éticas internacionales (5), el primero fue conocido y aprobado por Comité Ético de Investigación Clínica del Hospital de la Santa Cruz y San Pablo (Barcelona) y por las autoridades sanitarias españolas. Los sujetos firmaron un consentimiento informado y fueron gratificados económicamente por su colaboración.
Sujetos: Siete varones (V) jóvenes y sanos (edad: 22,75 ± 1,48 años, peso: 79,00 ± 8,76 kg y talla: 177,12 ± 8,18 cm), y ocho mujeres (M) jóvenes y sanas (edad: 22,37 ± 1,41 años, peso: 56,64 ± 4,83 kg. y talla: 162,75 ± 3,88 cm) fueron incluidos en este ensayo, tras obtener su consentimiento informado y resultados normales a partir de la exploración física y neuropsiquiátrica, así como de las evaluaciones bioquímicas y hematológicas. En caso de mujeres, se verificó que todas aplicaran medidas eficaces para la anticoncepción (únicamente, sujeto # 09 tomaba gestágenos orales) y que no estuvieran lactando ni embarazadas. Ningún sujeto superó un 28% de índice de masa corporal (6). Adicionalmente, se verificó que ninguno presentaba alguna de las siguientes condiciones: historia previa de alcoholismo o drogodependencia, consumidor importante de bebidas estimulantes (más de 5 porciones diarias de café, té, cola, chocolate, etc.), antecedentes de alergia, idiosincrasia o hipersensibilidad a fármacos, serología positiva para la hepatitis B o por virus HIV; antecedentes o presencia de patología renal, hepática, gastrointestinal, psiquiátrica u otras enfermedades crónicas que comprometan el estado general, antecedentes de cirugía mayor o enfermedad grave en los 3 meses anteriores al estudio, haber participado en el desarrollo de otro ensayo clínico durante el mes precedente al inicio de este estudio, ingesta de medicamentos psicoactivos u otros fármacos que pueden producir interacción con benzodiacepinas (teofilina y derivados, preparados vitamínicos con complejo B, anti-H1, nootrópicos y otros estimulantes cerebrales) durante las dos semanas previas, así como incapacidad para comunicarse o cooperar con los investigadores. No se aceptó el uso de medicación concomitante a lo largo del ensayo.
Condiciones Experimentales: Tras ayuno de 10 horas (hs) para sólidos y 6 hs para líquidos, a cada sujeto se le administró una dosis oral única de 2 mg de loracepam (LOR). A través de una bránula insertada en la vena antecubital hubo extracción sanguínea (8mL) a los siguientes tiempos: 0, 0,5h, 1h, 1,5hs, 2hs, 3hs, 4hs, 6hs, 7hs, 9hs, 10hs, 12hs, 24hs y 48hs postmedicación. Las muestras se depositaron y centrifugaron en tubos heparinizados, se separó el plasma, y se depositó en tubos plásticos y se congeló a -200C para su análisis posterior. Todos los participantes recibieron una merienda estandarizada a 2hs, 4hs, 6hs, 8hs y 10hs, se mantuvieron en reposo relativo y la ingesta de líquidos (agua) fue ad libitum a partir de 2hs. Se mantuvo una supervisión constante y se procedió con el control incruento de las constantes vitales en forma periódica. Puesto que los sujetos volvían al Servicio a 24hs y 48hs, se hicieron pruebas de laboratorio y gabinete tanto al iniciar como al finalizar cada sesión.
Cuantificación de Fármacos en Plasma: Se descongelaron alícuotas de plasma para proceder con extracción en fase sólida. Procesamiento de la muestra: Extracción: acondicionamiento 2 mL MeOH, 1 mL tampón K3PO4 0.1 M pH 6; plasma 1 mL + lavado: 1 mL tampón K3PO4 0.1 M pH 6 y 3 mL de acetonitrilo/H2O 20/80; elución: .0.5 mL MeOH/ácido acético 99.9/0.1 + 0.5 mL MeOH/ácido acético 99.9/0.1. El eluato se secó depositado en un evaporador de muestras durante 1.5hs a 30ºC. Condiciones cromatográficas: Solución de H2O/Acetonitrilo/MeOH 50/41/9 + 0.15 mL ácido perclórico para mantener pH 2.7-2.8, como fase móvil. Velocidad de flujo = 1 mL/min. Bomba Bromma LKB 2150®, columna Ultrasphere® C18 5u 150×4,6 mm, detector Waters 480-UV® para absorción UV con sensibilidad= 0.01 con lectura en 230 nm, integrador Merck Hitachi D-2500 Chromato-Integrator® para lectura cromatográfica y cuantificación del analito. Validación: Límite de cuantificación= 1.5 ng de LOR/mL de plasma. La linealidad se valoró con 5, 10, 15 20 y 30 ng/mL; coeficiente r= 0,9991; la exactitud y precisión se comprobó con concentraciones crecientes de 10 a 30 ng/mL, una recuperación creciente de 79.74 a 87.49% y un coeficiente de variación que osciló entre 1,20 y 4,09%.
Procesamiento de Datos y Análisis: Con las concentraciones plasmáticas (Cp) a cada tiempo de lectura se graficaron los perfiles para cada grupo según el sexo. De cada sujeto se identificó la concentración máxima (CMAX) y el tiempo de la CMAX (TMAX), y se calculó el tiempo de vida media de eliminación (t1/2) y el Área Bajo la Curva de 0-48 hs (ABC0-48) aplicando el programa PK-calc (7). Los datos se agruparon por sexo; se comparó las Cp a cada tiempo de lectura y los parámetros cinéticos aplicando test de t (prueba de Mann-Whitney para TMAX); y cualquier valor p<0,05 se consideró como significativo estadísticamente.
Resultados
El fármaco se detectó en plasma a partir de 1h. Los perfiles se muestran en la Figura 1, con una concentración media más elevada a 1.5 hs en mujeres (Cp= 17,08 ± 4,24 ng/nL, V= 14,69 ng/mL; M= 19,18 ng/mL, p= 0,033); los parámetros se describen en el Cuadro 1. La CMAX fue de 19,66 ng/mL (V= 16.28 ± 2,51 ng/mL, M= 23,05 6.12 ng/mL, p= 0.018), que se alcanzó a un TMAX de 1h para el 47% de los sujetos (n= 7; V= 3, M= 4), a 3 hs en el 27% (n= 4; V= 2, M= 2) y en pares a tiempos intermedios. El descenso progresivo de los niveles sanguíneos se correspondió con un t1/2 general de 11,33 hs (V= 11,98 ± 2.39 hs, M= 10.68 ± 2.69 hs y el fármaco resultó indetectable a 48 hs en todos los sujetos. El ABC0-48 tuvo una magnitud global de 199,46 ng/mL·h (168,36 ± 28,09 ng/mL·h, M= 230.57 ± 100,38 ng/mL·h.
Hubo una buena tolerabilidad sistémica al LOR; no se registraron cambios relevantes a nivel cardiovascular y hematológico, ni en las pruebas bioquímicas de control.
| | | |||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | ||||||
| | | | | | | | | | ||
| | | | | | | | ||||
* p<0.05; x = promedio, DE= desviación estándar.
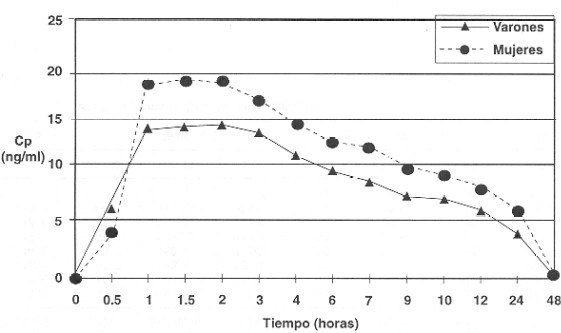
Figura 1. Perfil de las concentraciones plasmáticas de loracepam tras una dosis oral única de 2 mg en voluntarios sanos (7 varones y 8 mujeres), cuantificadas por HPLC.
Discusión
La pronta aparición del LOR en plasma luego de su administración oral, concuerda con la muy documentada rápida absorción del ansiolítico, aunque el tiempo registrado para la CMAX fue relativamente variable (entre 1 y 3 hs tras la ingesta). La dosis oral de 2 mg llevó a alcanzar una CMMAX global de 19.9 ng/mL, valor que se sitúa dentro del ámbito de CMAX informadas (17 - 27 ng/mL) tras la administración de una misma dosis (8-13). Además, LOR es una benzodiacepina descrita como de acción intermedia (t1/2 menor de 24 hs) debido a que su t1/2 de eliminación ronda las 14 hs con un ámbito que varía entre 9 y 35 hs (8-13), valor independiente de la dosis y vía de administración.
Al analizar los resultados agrupados por sexo, el perfil de las Cp resultó sistemáticamente más elevado en mujeres, lo que resultó concordante con un AUC notablemente mayor que no alcanzó significación estadística debido, probablemente, a la también mayor variabilidad dentro del grupo. Además, los valores de CMAX fueron claramente diferenciables en función del sexo, dado que la Cp significativamente mayor se obtuvo en el mismo grupo de mujeres. Al considerar tales disparidades en el comportamiento cinético del LOR, este fármaco parece merecer una evaluación más exhaustiva con muestras mixtas de mayor tamaño, en vez de suponerlas como producto del azar. Alternativamente, se ha documentado una cinética no diferencial en función del sexo para algunos fármacos(4,14-17), incluidos psicofármacos, lo cual favorece la generalización de sus esquemas posológicos.
Finalmente, los resultados obtenidos con este ensayo permiten concluir que efectivamente se señalan algunas evidencias que orientan hacia una diferenciación en la farmacocinética del LOR como consecuencia del factor sexo. Es importante destacar que, solo mediante la realización de este tipo de ensayos se puede establecer objetivamente la existencia real de posibles disparidades en el comportamiento cinético debido al sexo; y esto es relevante por cuanto, en la misma medida que tales diferencias no se establecen, se facilita el manejo terapéutico y la dosificación de los agentes farmacológicos.
Agradecimientos
Por su colaboración durante la etapa experimental, a la Dra. MC Bayes Genís y al personal del Área de Investigación Farmacológica, Instituto de Investigación- HSCSP, Barcelona.
Referencias
1. Levey BA.: Bridging the gender gap in research. Clin Pharmacol Ther 1991; 50: 641 - 646. [ Links ]
2. Rosser SV.: Gender bias in clinical research and the difference it makes. Appl Clin Trials 1993; 2: 44 - 50. [ Links ]
3. Yonkers KA, Harrison W.: The inclusion of women in psychophar-macologic trials. J Clin Psychopharmacol 1993; 13: 380 - 382. [ Links ]
4. Yonkers KA, Kando J, Cole JO, Blumenthal S.: Gender differences in human pharmacokinetics and pharmacodynamics of psychotropic medication. Am J Psychiatry 1992; 149: 587 - 595. [ Links ]
5. Gallant D, Krinsky SL.: Ethical and legal considerations in drug trials. En: Prien RF, Robinson DS (Eds.): Clinical evaluation of psychotropic drugs: principles and guidelines. New York: Raven Press, 1994: 261 - 280. [ Links ]
6. Lentner C. (Ed.): Body mass and height of adults. En: Geigy Scientific Tables, Somatometric Data, vol 3. Basle: International Medical and Phamaceutical Information Ciba-Geigy Limited, 1984: 324 - 329. [ Links ]
7. Shumaker RC.: PKcalc a basic interactive computer program for statistical and pharmacokinetic analysis of data. Drug Metab Rev 1986; 17: 331 - 348. [ Links ]
8. Greenblatt DJ, Shader RI, Franke K et al. Pharmacokinetics and bioavailability of intravenous, intramuscular and oral lorazepam in humans. J Pharmaceut Sci 1979; 68: 57 - 63. [ Links ]
9. Klotz U, Kangas L, Kanto J.: Clinical pharmacokinetics of benzodiazepines (lorazepam) Progress Pharmacol 1980; 3: 22 - 30. [ Links ]
10. Bourin M.: Pharmacocinétique des benzodiacépines. En: Les benzodiacépines, de la pharmacocinétique á la dependence, 2éme édition. Paris: Ellipses, 1989: 31 - 98. [ Links ]
11. Abernethy DR, Greenblatt DJ.: Drug disposition in obese humans. An update. Clin Pharmacokinet 1986; 11: 199 - 213. [ Links ]
12. Herrman RJ, VanPham JD, Szakacs CBN.: Disposition of lorazepam in humans beings: enterohepatic recirculation and first-pass effects. Clin Pharmacol Ther 1989; 46: 18 - 25. [ Links ]
13. Feely M, Pullar T.: Pharmacokinetic differences between benzodiazepines. En: Hindmarch I, Beaumont G, Brandon S, Leonard BE (Eds.): Benzodiazepines, current concepts. Chichester: John Wiley & Sons Ltd.; 1990; 61 – 72. [ Links ]
14. Kirkwood C, Moore A, Hayes P, DeVane L, Pelonero A.: Influence of menstrual cycle and gender on alprazolam pharmacokinetics. Clin Pharmacol Ther 1991; 50: 404 - 409. [ Links ]
15. Sáenz-Campos D, Bayés MC, Martín S et al.: Gender related pharmacokinetics od diltiazem in healthy subjects. Int J Clin Pharmacol Ther 1995; 33: 397 – 400. [ Links ]
16. Sáenz-Campos D, Bayés MC, Massana E et al. Sex-related pharmacokinetic and pharmacodynamic variation of lisinopril. Meth Find Exp Clin Pharmacol 1996; 18: 533 - 538. [ Links ]
17. Boni JP, DeCleene ShA, Cevallos WH, Hicks DR, Korth-Bradley JM.: Effects of age and grender on the pharmacokinetics of bromfenac in healthy volunteers. Ann Pharmacother 1997; 31: 400 – 405. [ Links ]
1. Departamento de Farmacología y Toxicología Clínica, Escuela de Medicina, Universidad de Costa Rica; Departamento de Farmacoterapia, Caja Costarricense de Seguro Social. Teléfono (506) 222-1878 y 256-4333; fax (506) 257-7004.
2. Laboratorio de Cromatografía, Área de Investigación Farmacológica, Instituto de Investigación, Hospital de la Santa Cruz y San Pablo (HSCSP), Barcelona.
3. Area de Investigación Farmacológica, Instituto de Investigación, HSCSP.
