











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
PermalinkIntroducción
El género Brycon (Characiformes: Characidae), consta de 42 especies y se distribuye desde el sur de México al Río La Plata en Argentina, con mayor diversidad en los ríos de Panamá y los ríos transandinos de Colombia y Ecuador (Silva, Oliveira, De Lima, & Matsumoto, 2008); este género comprende la especie endémica de Colombia Brycon henni (Eigenmann, 1913), conocida como "Sabaleta", presente en los principales afluentes de las cuencas de los ríos Cauca, Magdalena, San Juan, Dagua, Patía y San Jorge (Montoya, Carrillo, & Olivera, 2006a) y acuíferos de áreas productoras de café de la región central de Colombia. B. henni tiene una gran significación cultural en las zonas donde ocurre (Lenis, Cruz, & David, 2015), principalmente por la calidad de su carne y su comportamiento en la pesca deportiva (Pineda et al., 2007).
La especie ha sido estudiada desde el punto de vista biológico, anatómico y ecológico (Maldonado et al., 2005; Montoya et al., 2006b; Botero & Ramírez, 2011), productivo y reproductivo (Tabares, Ruíz, Arboleda, & Olivera, 2006; Tabares, Ruíz, Arboleda, & Olivera, 2007; Lenis, Restrepo, & Cruz, 2009; Chacón, Angel, Serna, Echavarría, & Vélez, 2015; Aguirre & Muñoz, 2015; Lenis et al., 2015), citogenético (David, Vásquez, Ruiz, & Olivera, 2008; Muñoz, Villamarín, & Londoño, 2016; De Miranda et al., 2014) y molecular, con marcadores moleculares RAPD (Pineda et al., 2007; Hurtado, Mancera, & Saldamando, 2011). A pesar de ello, el conocimiento sobre algunos aspectos aun es fragmentado y no hay estudios de variabilidad genética basados en marcadores moleculares codominantes, como los microsatélites, los cuales han sido ampliamente usados en otras especies de Brycon, como Brycon opalinus (Cuvier, 1819) por Barroso, Hilsdorf, Moreira, Cabellod, y Traub (2005), Brycon orbignyanus (Eigenmann & Norris, 1900) por Rodriguez et al. (2010); Lopera et al. (2010); Moreira, Polo, Silva, y Menezes (2015), Brycon insignis (Eigenmann & Norris, 1900) por Matsumoto y Silva (2009), Brycon hilarii (Valenciennes, 1850) por Sanches & Galetti (2012) y Brycon orthotaenia (Günther, 1864) por Sanches et al. (2012).
Los niveles de variabilidad genética de las poblaciones naturales de peces pueden ser afectados por características del ambiente en el que viven o bien pueden reflejar los eventos ambientales históricos que han sucedido en la región que ocupan (Palma, González, Romo, Ruiz, & Fuentealba, 2007). Según Mancera et al. (2013) las especies de peces en Colombia están sujetas a procesos de sobrepesca, introducción de especies exóticas, deterioro de hábitat y fragmentación de los ecosistemas, la cual limita el flujo genético entre las poblaciones. El monitoreo de la diversidad genética de las poblaciones naturales de peces es importante para su conservación; los marcadores moleculares son herramientas útiles para ello (Povh et al., 2008) y los microsatélites son los más ampliamente utilizados, especialmente en estudios genéticos de poblaciones de peces tropicales de agua dulce (Piorski et al., 2008), ya que permiten comparar entre poblaciones, dadas sus propiedades de abundancia, hipervariabilidad y herencia mendeliana (Ellegren, 2004; Kumar, Tomar, Kumar, & Singh, 2017).
En la cuenca alta del río Cauca, no se han realizado estudios sobre el estado de las poblaciones de B. henni que habitan en sus afluentes, no obstante, esta es una de las regiones de Colombia con mayor desarrollo de la agroindustria y la actividad minera; muchos de estos cauces son objeto de extracción de piedra y arena, y se han establecido represas para la generación hidroeléctrica y las actividades recreativas (CVC, 2004). Aunque B. henni no se encuentra en ninguna de las categorías de riesgo propuestas por el libro rojo de peces dulceacuícolas de Colombia (Mojica, Usma, Álvarez-León, & Lasso, 2012), la alteración de su hábitat y el aislamiento de las poblaciones por el deterioro de la calidad del agua del río Cauca pueden tener un efecto sobre la estructura genética de las poblaciones, con consecuencias sobre su conservación (Hurtado et al., 2011). Este estudio tuvo como objetivo analizar la estructura genética poblacional de B. henni en cuatro cuencas de la región Andina colombiana, utilizando marcadores moleculares microsatélites e identificar las poblaciones con prioridad de conservación.
MATERIALES Y MÉTODOS
Recolección de muestras: Se obtuvieron 60 individuos de B. henni, distribuidos en cuatro cuencas hidrográficas de la región Andina de Colombia (quince en cada una). Estas fueron los ríos Risaralda (RR) (05°06´ N - 75°50´ W), Campoalegre (RC) (4°56´ N - 75°37´ W), Riofrío (RRf) (04°07´ N - 76°21´ W) y Río Chinchiná (RCh) (5°03´ N - 75°36´ W). Los individuos fueron capturados con métodos de pesca artesanal (atarraya) y la especie fue determinada de acuerdo a Maldonado et al. (2005).
Procedimientos de laboratorio: 100 mg de músculo esquelético de los animales muestreados se almacenaron en etanol al 95 % hasta su uso. El ADN genómico se aisló usando el protocolo de extracción con Fenol-Cloroformo, de acuerdo con Taggart, Hynes, Prodohl & Fergusson (1992) modificando el tipo de enzima proteolítica y el detergente iónico, para ello se resuspendieron 100 mg de musculo esquelético en 375 µl de EDTA (ácido etilendiamino tetraacético) 0.2 M, 1 % de SDS y 7 µL de proteinasa K (200 µg/ml), seguida por incubación a 37 °C por 15 h. Posteriormente se adicionaron 10 µl de DNAsa (2 mg/ml) y se incubaron a 37 °C por 1 h. Se adicionaron 400 µl de fenol (pH 8.0), con agitación vigorosa por 20 s y luego más suavemente por 10 min; se adicionaron 400 µl de alcohol cloroformo: isoamilico (24:1) a cada tubo, y se realizó la misma agitación seguida de centrifugación a 12 000 g por 3 min. Luego el sobrenadante fue mezclado por inversión de 5 a 6 veces con etanol frio al 99 % por 15 min, seguido por un lavado con etanol al 70 % por 1h con mezclado suave. Finalmente se decantó el etanol y se dejó secar el ADN por 10 min a temperatura ambiente y se resuspendió en buffer TE (10 mM Tris, 1 mM EDTA; pH 8.0).
El ADN fue cuantificado en un nanodrop 2000 Thermo Sientific y su integridad verificada con electroforesis horizontal en un gel de agarosa a una concentración del 1 %. Se utilizaron doce cebadores (Tabla 1) previamente evaluados en otras especies de Brycon: Bh17, Bh5, Bh6, Bh8, Bh15 (Sanches & Galetti, 2006), BoM1, BoM2, BoM5, BoM6, BoM7, BoM12 y BoM13 (Barroso et al., 2003). Su síntesis, amplificación y secuenciación fue realizada mediante un secuenciador ABI3730XL (Macrogen Sequencing Service, Corea) y el tamaño de los alelos se midió mediante el software Peak scanner ™ V.1.0.
TABLA 1: Información de los microsatélites empleados en este estudio. Brycon opalinus (Bom1, Bom2, Bom5, Bom6, Bom7, Bom12, Bom13) y Brycon hilarii (Bh5, Bh6, Bh8, Bh15, Bh17) TABLE 1: Microsatellites information used in this study. Brycon opalinus (Bom1, Bom2, Bom5, Bom6, Bom7, Bom12, Bom13) and Brycon hilarii (Bh5, Bh6, Bh8, Bh15, Bh17)
| Locus | Secuencia del cebador (5`- 3`) | Motivo- repetición | Rango alélico (pb) | |
| - | - | - | Reportado (pb) | Encontrado (pb) |
| Bh17 | F: GTCAGCACTCAGCACATAGC R: AGAGAGCCTGAAAGTGAGTC | (GTTT)4 (GGTTT)3 | 152-212 | 150-225 |
| Bh5 | F: CTTCCACTCATACCGGCACT R: ACATCTGGCATTAGGCATAG | (AC)13 | 204-220 | 164-226 |
| Bh6 | F: GCGTTGCGTGTGTATGTTAA R: AGAGGTGTCCACAAAGTTTT | (GT)14 | 160-184 | 168-193 |
| Bom1 | F: CCATCTCTACTTTTTGGTTCC R: TGCCCAGATACAGCCC | (CA)16 | 139-173 | 127-212 |
| Bom12 | F: GCAGCAGAAAGAAACAG R: CGGGGAGATTTCAACCT | (GT)15 | 78-118 | 55-118 |
| Bom13 | F: CATTTCCTCAGTCCTTTTCAGC R: CCCACTTAGGGTCGCAC | (CT)11 | 154-176 | 158-192 |
| Bom2 | F: CTGGGCAGCGGAAGAG R: CCCACATCTCTCCCCTTCG | (CA)45 | 162-242 | 216-240 |
| Bom5 | F: CGACCACAATAGGATTAGGG R: CTGGAGTTTGTGTGTGGA | (AC)4 T(AC)10 AT(AC)5 | 117-151 | 129-165 |
| Bom6 | F: GGAGTTTGTGTGTGGAGACCGAG R: GCACGCAGACACCAGA | (CA)5 TA(CA)10 T(CA)4 | 142-178 | 125-188 |
| Bom7 | F: CTCTGCCCCAGGTCTCACT R: CGGGAGTGACGAAATG | (CA)31 | 172-236 | 164-225 |
Informatividad de los microsatélites y características de las poblaciones: Los marcadores empleados en el presente estudio fueron seleccionados por haber sido los más polimórficos en investigaciones previas con otras especies de Brycon (Sanches & Galetti, 2006; Matsumoto & Silva, 2009). Rodríguez et al., (2010), Sanches y Galetti (2012), Lopera et al., (2010) y Sanches et al. (2012), que evaluaron 5, 7, 4 y 5 loci en las especies B. opalinus, B. hilarii, B. orbignyanus y B. orthotaenia, respectivamente. Los microsatélites BoM2 y BoM7 han mostrado alto polimorfismo en estudios realizados en B. opalinus, con 31 y 25 alelos respectivamente (Barroso et al., 2003).
Los marcadores fueron descritos mediante la estimación de parámetros como el número de alelos (NA), el número promedio y efectivo de alelos (NPA y NEA), la heterocigosidad observada (HO) y esperada (HE), el coeficiente de endogamia (FIS), las desviaciones del equilibrio Hardy Weinberg (EHW) dentro de las poblaciones y número privado de alelos para cada población (NPA) con el programa GenAlex (Peakall & Smouse, 2006). Se estimó la riqueza alélica (RA) utilizando el programa FSTAT (Goudet, 2002) y el contenido de información polimórfica (PIC) con la aplicación Microsatellite Toolkit para Excel (Park, 2001).
Estructura genética: Fue analizada mediante los estadísticos de estructura poblacional de Wright (FIT, FST y FIS) y el AMOVA basado en el estadístico RST asumiendo un modelo de mutación por pasos (SMM), que refleja más exactamente el patrón de mutación de los microsatélites (Balloux & Lugon-Moulin, 2002), así como la prueba de desequilibrio de ligamiento con 10 000 permutaciones, mediante el programa Arlequin (Excoffier, Laval & Scheneider, 2005). Las distancias genéticas entre las poblaciones se calcularon como Distancia de Nei (Da) (Nei, 1972) y se construyó un dendrograma basado en la UPGMA (Unweighted Pair Group Method with Arithmetic mean) con TFPGA (Miller, 1997), para representar gráficamente esas distancias. Por último, se utilizó el programa Structure (Pritchard, Stephens, & Donnelly, 2000) para asignar individuos a los grupos por similitud genética, mediante métodos bayesianos. Se tomaron diferentes números de poblaciones, asumiendo el modelo de mezcla y las frecuencias alélicas correlacionadas, con 100 000 a 500 000 iteraciones y dos repeticiones para cada K, cuyo valor más probable fue determinado por el método de Evanno, Regnaut, y Goudet (2005), con el Software Structure Harvester (Earl & VonHoldt, 2012).
RESULTADOS
Informatividad de microsatélites: Dos de los 12 marcadores seleccionados, Bh8 y Bh15, no amplificaron exitosamente. Las características de los 10 loci microsatélites analizados se presentan en la tabla 2.
TABLA 2: Resumen estadístico para los loci microsatélites analizados TABLE 2: Stats of microsatellites loci analyzed
| Locus | NA | NPA | NEA | RA | PIC | HE | HO | Estadísticos F | r | EHW | ||
| - | - | - | - | - | - | - | - | FIS | FIT | FST | - | - |
| Bh17 | 11 | 5.5 | 2.581 | 5.291 | 0.655 | 0.572 | 0.339 | 0.407 | 0.503 | 0.162 | 0.148 | 3 |
| Bh5 | 10 | 5.2 | 3.450 | 5.277 | 0.701 | 0.703 | 0.354 | 0.497 | 0.520 | 0.046 | 0.205 | 3 |
| Bh6 | 9 | 5.5 | 3.891 | 5.358 | 0.750 | 0.741 | 0.533 | 0.280 | 0.318 | 0.053 | 0.119 | 2 |
| BoM1 | 20 | 8.2 | 3.945 | 6.512 | 0.720 | 0.701 | 0.350 | 0.501 | 0.531 | 0.061 | 0.206 | 3 |
| BoM12 | 15 | 5.0 | 1.737 | 3.959 | 0.387 | 0.347 | 0.200 | 0.424 | 0.501 | 0.133 | 0.109 | 3 |
| BoM13 | 12 | 6.5 | 2.776 | 5.511 | 0.609 | 0.572 | 0.433 | 0.242 | 0.312 | 0.093 | 0.088 | 3 |
| BoM2 | 20 | 8.2 | 2.674 | 6.267 | 0.640 | 0.555 | 0.370 | 0.332 | 0.431 | 0.148 | 0.119 | 3 |
| BoM5 | 15 | 7.5 | 3.692 | 5.934 | 0.722 | 0.703 | 0.250 | 0.644 | 0.666 | 0.061 | 0.266 | 4 |
| BoM6 | 8 | 4.0 | 2.232 | 3.747 | 0.513 | 0.510 | 0.142 | 0.722 | 0.756 | 0.123 | 0.244 | 4 |
| BoM7 | 16 | 8.7 | 4.135 | 6.986 | 0.785 | 0.739 | 0.402 | 0.456 | 0.500 | 0.081 | 0.194 | 4 |
Número de alelos (NA), Número promedio de alelos (NPA), Número efectivo de alelos (NEA), Riqueza alélica (RA), Contenido de información polimórfica (PIC), Heterocigosidad esperada (HE), Heterocigosidad observada (HO), Coeficiente de endogamia respecto a la subpoblación (FIS), coeficiente de endogamia total (FIT), Indice de diferenciación genética entre subpoblaciones (FST), Frecuencia de alelos nulos (r) y poblaciones desviadas del equilibrio Hardy-Weinberg (EHW) por locus.
Alleles number (NA), Average number of alleles (NPA), Effective number of alleles (NEA), Alelic Richness (RA), Polymorphic information contain (PIC), expected heterozygosity (HE), observed heterozygosity (HO), Coefficient of inbreeding with respect to the subpopulation (FIS), Total coefficient of inbreeding (FIT), genetic differentiation index between subpopulations (FST), Null aleles frequency (r) y populations deviated from Hardy-Weinberg equilibrium (EHW) by locus.
Todos los loci fueron polimórficos, el valor más alto de NA se presentó en BoM1, BoM2 y BoM7, con 20, 20 y 16 alelos totales, respectivamente. El NPA general fue de 6.45, con valores por cada locus que van desde 4 a 8.75. Todos los marcadores fueron informativos y variables, excepto BoM12, con PIC de 0.39, considerado razonablemente informativo (Botstein, White, Skolnick, & Davis, 1980) y una HE inferior a 0.5. La RA, que estima la diversidad alélica de una población independientemente del número de muestras analizadas, osciló entre 3.75 y 6.99.
Todos los marcadores mostraron déficit de heterocigotos. Se presentaron valores de FIS entre 0.242 y 0.722; el índice de fijación genética FIT mostró una fuerte desviación respecto a la situación de panmixia (0.504) y se evidenció una desviación significativa de EHW (P < 0.05), dada posiblemente por la probabilidad de alelos nulos, que en los loci analizados se encuentra entre 0.088 y 0.26. De los diez loci analizados, cuatro presentaron alta probabilidad de ocurrencia de alelos nulos, con valores de r > 0.2 (Bh5, BoM1, BoM5, y BoM6), mientras que la probabilidad en los loci restantes fue moderada, con valores de 0.05 < r < 0.2.
Se estimaron las desviaciones del desequilibrio de ligamiento para evaluar la independencia de genotipos entre los loci, pues la presencia de endogamia en una población generaría correlación entre todos los loci (desequilibrio por ligamiento) (Gastric, Bernatchez, Belkhir, & Bonhome, 2002). Se encontraron desequilibrios de ligamiento (DL) significativos (P < 0.05) entre algunas parejas de marcadores; los marcadores que presentaron mayor DL en todas sus comparaciones fueron el Bh17 y el Bh5. La población que presentó mayor número de pares de marcadores en DL, fueron RRf y RCh, con 31 y 28 parejas de marcadores respectivamente, mientras que RR fue la población que menos marcadores en desequilibrio mostró, con seis parejas.
Variación genética: Se encontraron un total de 136 alelos en todas las poblaciones. La población de la cuenca del Río Campoalegre (RC) presentó los mayores NA (81), NPA (8), AP (34) y HE de 0.689 ± 0.05, mientras que la población de la cuenca del Río Riofrío (RRf) presentó los valores más bajos, con 53, 5 y 10 alelos respectivamente y HE de 0.515 ± 0.055 (Tabla 3).
TABLA 3: Parámetros de diversidad genética en las poblaciones de B. henni analizadas TABLE 3: Genetic diversity parameters in B. henni populations
| Población | NPA | AP | HE (D.E.) | HO (D.E.) | FIS |
| Río Campoalegre (RC) | 8 | 34 | 0.689 (0.050) | 0.439 (0.062) | 0.368 |
| Río Risaralda (RR) | 7 | 13 | 0.628 (0.072) | 0.399 (0.080) | 0.434 |
| Río Chinchiná (RCh) | 6 | 19 | 0.625 (0.035) | 0.272 (0.043) | 0.551 |
| Río Riofrío (RRf) | 5 | 10 | 0.515 (0.055) | 0.241 (0.030) | 0.479 |
Se encontró un total de 78 % de alelos con frecuencia menor a 10 %; se puede inferir la pérdida de variabilidad genética debido a la disminución de los alelos. Todas las poblaciones presentaron déficit de heterocigotos. La mayor diferencia entre ambas heterocigosidades se presentó en RCh, con FIS de 0.551 y la menor en RC, con FIS de 0.368.
Estructura genética: De acuerdo con los valores de FIS y FIT (0.451 y 0.504 respectivamente), los cuatro grupos analizados tienen 45.1 % de déficit de heterocigotos y teniendo en cuenta la población total, el déficit es de 50.4 %. El AMOVA, basado en el estadístico RST, detecto diferencias significativas (P < 0.05) y reveló que 90 % de la variabilidad total encontrada se explicaba por variabilidad entre individuos dentro de poblaciones. Por su parte, la diferenciación entre poblaciones, basada en el estadístico FST, que expresa la reducción promedio en la heterocigosidad de una subpoblación generada por deriva genética, presentó un valor moderado y altamente significativo (0.09), por lo que solo el 9 % de la varianza es atribuida a diferencias entre las subpoblaciones, por lo que se asume algún flujo genético entre ellas.
Los valores de FST por parejas de poblaciones (Tabla 4), oscilaron entre 0.058 y 0.081 y todos fueron altamente significativos (P < 0.001), excepto entre RCh y RRf, con un valor bajo y no significativo, lo que podría deberse a algún programa de repoblación, aunque no hay documentación publicada al respecto. Finalmente, de acuerdo con la distancia genética de Nei (1972), la población más divergente fue RC, mientras que RCh y RRf fueron las más cercanas genéticamente entre ellas, como se muestra en la tabla 4. Aunque los valores de significancia del dendrograma fueron inferiores a 50 %, corresponden con los obtenidos con el estadístico FST, que fueron en su mayoría significativos.
TABLA 4: Distancias genéticas por parejas de poblaciones basadas en el parametro FST TABLE 4: Genetic distances by population’s pair base don FST parameter
| RC | RR | RCh | RRf | |
| RC | - | - | - | - |
| RR | 0.058** | - | - | - |
| RCh | 0.074** | 0.066** | - | - |
| RRf | 0.081** | 0.066** | 0.031NS | - |
**P < 0.001; *P < 0.05
En la Fig. 1 se representa gráficamente la estructura de la población (Pritchard et al., 2000) (Fig. 1.). Cada línea vertical representa un individuo que está dividido hasta en cuatro colores, según el número de k “Clusters”. Los colores en cada individuo representan la proporción estimada de pertenencia a cada “Cluster”. Se encontró la tendencia a cuatro poblaciones ancestrales. Según el coeficiente de pertenencia, las poblaciones son heterogéneas (Tabla 5). En RCh y RRf, cerca de 50 % de los individuos provienen del mismo grupo (rojo), mientras que las poblaciones RR y RC tienen mayor heterogeneidad con respecto a posibles poblaciones ancestrales.
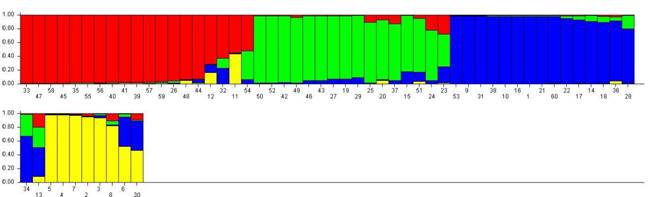
Fig. 1. Fig. 1 Representación gráfica de estructura de la población. Cada línea vertical es un individuo y cada color representa un “cluster”. La distribución de los colores representa la pertenencia de cada individuo a un grupo determinado. Individuos: 2-18 (RC), 19-36 (RR), 38-54 (RCh) y 55-72 (RRf). Graphic representation of population structure. Each vertical line is an individual and each color represents a "cluster". The distribution of the colors represents the belonging of each individual to a certain group. Individuals: 2-18 (RC), 19-36 (RR), 38-54 (RCh) and 55-72 (RRf).
TABLA 5: Proporción de membresía de cada población en los cuatro “Clusters” identificados en el análisis bayesiano TABLE 5: Membership proportion of each population in four clusters identified in Bayesian Analysis
| Población | Cluster | - | - | - |
| - | 1 (azul) | 2 (amarillo) | 3 (rojo) | 4 (verde) |
| RC | 0.353 | 0.447 | 0.109 | 0.091 |
| RR | 0.433 | 0.031 | 0.132 | 0.404 |
| RCh | 0.260 | 0.007 | 0.512 | 0.220 |
| RRf | 0.166 | 0.014 | 0.504 | 0.316 |
DISCUSIÓN
Las poblaciones de Brycon henni evaluadas en este estudio presentan una diferenciación genética moderada, posiblemente asociada al aislamiento físico actual entre ellas. Sin embargo, la menor distancia genética entre las poblaciones de RCh y RRf muestra cierta tendencia a pertenecer a un grupo común y la posible existencia de flujo génico entre ellas, posiblemente por influencia antrópica. Adicionalmente, el déficit de heterocigotos y el DL en muchos de los loci analizados sugieren adicionalmente la influencia de endogamia, o muestreo de individuos emparentados, principalmente en las poblaciones de RCh y RRf.
Las amplias diferencias entre los valores de NA y NEA permiten inferir que muchos de los alelos encontrados para estas poblaciones son considerados como alelos raros, esto se confirma con el alto PIC encontrado en todos los marcadores, excepto en uno de ellos, dado que son interespecíficos y fueron seleccionados a partir de los más polimórficos empleados en estudios previos en el género Brycon. Por su parte, se presentó menor HO frente a otras especies de Brycon, como B. orthotaenia (0.65 - 0.73) según Sanches et al. (2012) y B. hilarii (0.59 - 0.61) según Sanches y Galetti (2012).
El alto FIT evidencia un marcado déficit de heterocigotos con respecto a la población total, una de las causas de ello puede ser la aparición de alelos nulos, confirmada por los valores moderados a altos de probabilidad de presencia en todos los loci, o a la reducción de los tamaños poblacionales poblaciones debido al deterioro ambiental. Según Eguiarte, Aguirre, Scheinvar, González, y Souza (2010), este coeficiente señala el efecto conjunto de la endogamia (FIS) y la deriva genética en la diferenciación de las poblaciones (FST). Esta situación podría estar asociada al deterioro ambiental de las cuencas estudiadas, las cuales puede reducir las poblaciones de peces (Hurtado et al., 2011).
Aunque las poblaciones de B. henni estudiadas tienen algún nivel de diferenciación, el análisis sugiere que podría existir una tasa de migración considerable (2.86), que evidencia de un moderado flujo genético que sobrepasa los efectos de deriva e impide una mayor diferenciación local (Slatkin, 1994). Según Jiménez, Román-Valencia, & Cardona (1998), la especie presenta movimientos migratorios entre el cauce principal y todos sus tributarios, los cuales pueden obedecer a la búsqueda de mejores condiciones del agua (temperatura, transparencia). Aunque estas migraciones no están necesariamente asociadas a la reproducción (Perdomo, 1978), por lo que la construcción de embalses y el deterioro de la calidad de agua de los corredores fluviales puede afectar sus migraciones. Las cuencas de los ríos Risaralda, Riofrío, y Chinchiná, son receptoras de aguas residuales de una gran cantidad de centros urbanos y el agua contiene una alta carga de contaminantes como metales pesados y residuos de insumos agrícolas (CVC, 2004) que podrían estar afectando las poblaciones naturales de B. henni.
Además de la presencia de alelos nulos, el exceso de homocigotos también está definido por efectos de endogamia al interior de las poblaciones, dado el alto FIS, o, de una forma menos probable, por una mezcla de individuos de varias unidades panmícticas, con frecuencias alélicas diferentes (efecto Wahlund), que provoca un aumento de genotipos homocigotos, en las poblaciones analizadas (Pardo, Morales, & Cavadia, 2014) y que en el presente estudio pudo producirse como un artefacto de muestreo por realizar trampeos en diferentes sitios de los ríos con la consecuente toma de animales de diferentes agrupaciones genéticas. Adicionalmente, con la técnica empleada, la presencia de alelos nulos se debe generalmente a mutaciones en las secuencias que flanquean la región microsatélite, las cuales sirven como sitio de unión a los partidores diseñados para producir la amplificación por PCR, por lo cual un genotipo heterocigoto puede ser registrado erróneamente como homocigoto (Pemberton, Slate, Bancroft & Barrett, 1995).
Los valores de FIS encontrados en el presente estudio son superiores a los encontrados en otras especies de Brycon, como B. orbignyanus en Brasil, con -0.116 a -0.242 (Lopera et al., 2010) y -0.489 a -0.224 (Rodriguez et al., 2010), B. hilarii, con 0.096 a -0.109 (Sanches & Galetti 2012) y B. orthotaenia, de 0.12 (Sanches et al., 2012), pero similares a los datos reportados por Barroso et al., (2005) para poblaciones de B. opalinus de Río Preto, Brasil, con 0.148 a 0.414. En cuanto al NPA encontrado en este estudio, se obtuvieron datos semejantes al determinado por Sanches et al. (2012) en B. orthotaenia en el río San Francisco, en Brasil (5.6 a 7.2 alelos) y la RA encontrada presentó rango de valores más amplio que los hallados en especies como B. orthotaenia y B. hilarii, de 5.48 a 6.44 y 5.20 a 5.66, respectivamente (Sanches et al., 2012, Sanches, & Galetti, 2012).
La diversidad genética de muchas poblaciones de peces es muy variable, dependiendo de la especie, la ubicación del río y las presiones en cada ambiente (Povh et al., 2008). La mayor variabilidad genética se encontró en el RC, cuenca con el mayor número de AP y población más diferenciada; esta cuenca tiene aproximadamente 46.9 % de su territorio bajo alguna categoría de conservación y las comunidades que rodean el río lo han protegido, evitando que personas foráneas realicen actividades pesqueras (Castaño & Carranza, 2015).
Por otra parte, los valores más bajos de variabilidad genética en las otras poblaciones pueden ser consecuencia además, de la deposición de agua residual urbana e industrial y a la sobrepesca de la especie en dichos ríos, tal como lo sugirió Pineda et al., (2007) y Hurtado et al., (2011) en cuencas hidrográficas del departamento de Antioquia. Estos autores concluyeron que los efectos antrópicos de la sobrepesca, construcción de presas, deforestación y contaminación del agua, han contribuido al aislamiento de ciertos grupos, generando un alto nivel de estructura genética y un 50 - 70 % de polimorfismo. Los resultados actuales mostraron que las poblaciones de B. henni evaluadas tienen un nivel moderado pero altamente significativo de estructura genética con valores de diversidad genética que lo convierten en un valioso recurso que debe ser preservado. Actualmente no hay informes oficiales de monitoreo del estado de las poblaciones de peces en estas cuencas; sin embargo, los resultados sugieren que las poblaciones pueden estar alcanzando valores críticos de baja densidad, lo cual pone en riesgo su conservación.

