











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
PermalinkIntroduction
Phaseolus vulgaris is cultivated and consumed primarily in Latin America, Africa and Asia (Gepts et al., 2008). Several attempts have been made to transform the species to obtain traits that otherwise cannot be achieved by traditional crosses (Amugune, Anyango, & Mukiama, 2011). Some genetic manipulations have been carried out with biolistic but at low efficiencies (0.03-0.9 %) (Russell, Wallace, Bathe, Martinell, & McCabe, 1993; Aragão et al., 1996; Vianna et al., 2004).
Transgenic plants of P. vulgaris have been obtained with resistance to bean golden mosaic virus (BGMV) and with increased tolerance to herbicide ammonium gluphosinate Imazapyt by the biolistic approach (Aragão, Vianna, Albino, & Rech, 2002; Rech, Vianna, & Aragão, 2008; Aragão, Nogueira, Tinoco, & Faria, 2013). Transformed plants with the mutant gene associated to the Rep protein have shown a partial resistance to the BGMV and this trait has been then transferred to four commercial cultivars by traditional crosses (Faria et al., 2006). Furthermore a BGMV (EMBRAPA 5.1) was resistant line made by transforming with a genetic silence system against the viral gene rep (AC1) (Bonfim, Faria, Nogueira, Mendes, & Aragão, 2007; Aragão & Faria, 2009; Aragão et al., 2013). However, such transformation has a low efficiency and higher cost compared to the use of Agrobacterium-mediated transformation (Amugune et al., 2011).
There have been several efforts to conduct genetic transformation via A. tumefaciens (Franklin, Trieu, Cassidy, Dixon, & Nelson, 1993; Zhang, Coyne, & Mitra, 1997), with the efficiency of 2.8- to 28.6 % (Espinosa-Huerta, Quintero-Jiménez, Cabrera-Becerra, & Mora-Avilés, 2013; Collado et al., 2015). Regeneration of plants from transformed explants is difficult in genetic transformation protocols, and for recalcitrant species like common bean is one of the unsolved aspects (Liu, Park, Kanno, & Kameya, 2005; Amugune et al., 2011; Collado et al., 2015). Recently the green nodular compact calli were used as explants to obtain transformed plants, based on the indirect organogenesis regeneration pathway (Collado et al., 2015; Collado et al., 2016).
An efficient system for transformation remains elusive for Phaseolus vulgaris. Main goal of this study was to develop a transformation protocol for Phaseolus vulgaris var. Brunca via Agrobacterium tumefaciens considering several factors like bacterial concentration, incubation of the pre-culture and co-cultivation, antibiotics used and lethal concentrations of selective agents.
Material and methods
Plant materials: Experiments were performed from 2012 to 2015 using mature common bean seeds var. Brunca (its color black) donated by the Fabio Baudrit Moreno Agricultural Experimental Station [Estación Experimental Agrícola Fabio Baudrit Moreno (EEAFBM)] of the University of Costa Rica, located in La Garita de Alajuela, Costa Rica.
Seeds were firstly surface-sterilized according to Solís-Ramos et al. (2015). Embryonic axes (EA) were extracted with cutters, tweezers and stereoscopic microscope. The samples were surface-sterilized a second time with a solution of 0.1 % of bleach (commercial sodium hypochlorite) for 10 min and subsequently washed three times with sterile distilled water. Finally, EA were placed in the regeneration medium (RM) in petri dishes. For experiments whose pre-cultivation periods were considered, embryos were extracted at the moment of bacterial immersion by removing cotyledons and radicles.
Regeneration medium: Murashige & Skoog (1962) (MS) medium added with 1 mg/L BAP, 30 g/L sucrose, and 6 g/L agar was used (Solís-Ramos et al., 2015). The pH was adjusted to 5.7 prior to autoclave sterilization (120 °C, 15 kg/cm2, for 20 min). The cultures were incubated at 26 ± 1 °C with a photoperiod of 12 h light (30 μmol/m2s) and 12 h darkness.
Activation of bacterial strains: Agrobacterium LBA4404-ElectroMAX® cells (transformed with bifunctional BinRD29A or pCAMBIA1301 plasmids) were cultured in 25 mL of liquid Luria-Bertani (LB) medium (10 g/L triptone, 5 g/L NaCl, 5 g/L yeast extract) containing the appropriate antibiotics and allowed to grow for 48 h at 28 °C with 160 rpm agitation according to Solís-Ramos, González-Estrada, Nahuath-Dzib, Zapata-Rodriguez, & Castaño (2009). Subsequently, 200 µl of the bacterial suspension were inoculated in 25 mL of fresh LB medium containing the required antibiotics. The culture was allowed to grow for 24 h at 28 °C and 160 rpm agitation. Then, 25 mL of LB and 100 µM of acetosyringone (4’-hydroxy-3’,5’-dimethoxyacetophenone) were added to the 24-h culture which was incubated for 4 hr at 28 °C and 160 rpm agitation. The bacterial suspension was centrifuged at 5 000 rpm during 5 min at room temperature and resuspended in 20 mL of MS liquid media containing 1 mg/L BAP and 100 µM of acetosyringone until it reached 0.5 or 0.8 OD600nm depending on the treatment. The inocultion was performed by immersing the explants in the bacterial suspension for 15 min under constant agitation (900 rpm), and bacterial suspension excess was eliminated with sterile towel paper. The co-cultivation was performed in Petri dishes with regeneration medium under complete darkness at 25±1 °C and co-cultivation time depended on the treatments of each experiment.
Bacteria elimination after co-cultivation: The Agrobacterium elimination was performed after the co-cultivation in pre-cultivated or non-pre-cultivated explants. Explants were shaken at 90 rpm for 15 min in the bacterial suspension and then co-cultivated for four days. After co-cultivation, explants were washed three times with sterile distilled water for 5-10 min (manual agitation) with subsequent vacuum infiltration at 1.25 plg Hg during 30-40 min with solutions containing timemtin, cefotaxime or a mixture of both. The following treatments were evaluated: (T1) non-infected EA on antibiotic-free RM, (T2) co-cultivated EA on antibiotic-free RM, (T3) cocultivated EA on RM containing 500 mg/L timentin and 500 mg/L cefotaxime, (T4) co-cultivated EA on RM containing 500 mg/L timentin, and (T5) co-cultivated EA on RM containing 750 mg/L timentin.
EAs water excess was adsorbed with sterile paper towels before they were cultured in the RM supplemented with the selected treatment. Then, they were cultured under the conditions described previously. Twelve explants per treatment were used and the bacterial overgrowth (expressed as percentage) after seven days of cultivation was calculated. The best treatment to eliminate bacteria was selected and its effect on the in vitro regeneration was assessed.
Assessment of kanamycin (Kn) as selective agent: The explants with a pre-cultivation time of seven days were cultured in RM supplemented with 0, 25, 50, 100, 200 and 300 mg/L of kanamycin. Each treatment was replicated three times with seven explants per treatment. The explants were cultured at 25 °C under a 16 hr light/8 hr dark photoperiod. Sub-culture was performed every three weeks to the same medium. After three and six weeks of culture we recorded the survival rate, as well as the number of shoots and/or roots observed per explant.
Transient transformation experiments: The A. tumefaciens LBA4404-ElectroMAX® strain carrying the binary vector pCAMBIA1301 (11837 bp) was used. This vector contains the β-glucuronidase (gusA) reporter gene of Escherichia coli with an intron. The gusA gene under the 35S promoter of the cauliflower mosaic virus (CaMV) and the poly(A) ending sequence contains the gene hpt (hygromycin phosphotransferase, a selective marker gene) controlled by the 35S promoter and the poly (A) 35S CaMV terminator.
Non-precultured EAs were incubated at room temperature in the bacterial suspension under 0.5 OD600nm. Later, bacterial excess was adsorbed with sterile filter paper and EAs were placed in Petri dishes containing RM. The effect of the co-culture period was assessed under three, four, and five days of incubation at 25 °C in the dark (Table 1). A second experiment assessed a pre-culture time of two days and a co-culture time of four days (Table 1).
Table 1 Conditions of the genetic transformation of bean with LBA4404-ElectroMAX® of Agrobacterium
| Experiment | Pre-cultivation period (Days) | Number of explants | Plasmid | Infection | Co-cultivation period (Days) |
| 1 | 0 | 11 | 3 | ||
| 11 | pCAMBIA1301 | Immersion and agitation for 15 min. | 4 | ||
| 11 | 5 | ||||
| 2 | 2 | 197 | pCAMBIA1301 | Vacuum infiltration, 1 h, 1.25 plg Hg | 4 |
| 3 | 12 | 30 | pCAMBIA1301 | Immersion and agitation for 15 min | 3 |
| 4 | 2 | 405 | pBinRD29A-bifuncional | Vacuum infiltration, 1 h, 1.25 plg Hg | 4 |
| 5 | 8 | 60 | pBinRD29A-bifuncional | Vacuum infiltration, 1 h, 1.25 plg Hg | 4 |
| 6 | 10 | 120 | pBinRD29A-bifuncional | Vacuum infiltration, 1 h, 1.25 plg Hg | 4 |
| 7 | 12 | 30 | pBinRD29A-bifuncional | Immersion and agitation for 15 min | 3 |
| 8 | 17 | 45 | pBinRD29A-bifuncional | Vacuum infiltration, 1 h, 1.25 plg Hg | 4 |
Histochemical staining: After bacterial elimination, the gusA activity on EAs was detected by histochemical staining (Jefferson, Kavanagh, & Bevan, 1987). Explants were incubated overnight at 37 °C in 100 mM of phosphate sodium (NaH2PO4, pH 7.0) buffer containing 10 mM EDTA, 0.5 mM potassium ferrocyanide K3Fe (CN6), 0.5 mM potassium ferrocyanide K3Fe (CN)6∙3H2O, and 0.2 % Triton X-100. X-Gluc (5-bromo-4-chloro-3-indolyl-β-D-glucuronide) dissolved in dimethyl sulfoxide (DMSO) under a final concentration of 0.7 mg/L was added to the solution. After incubation, the staining solution was removed and EAs were put in ethanol (70 %) for 24 h to eliminate chlorophylls. The presence of blue points was identified by means of a stereomicroscope (LEICA E24HD) and was registered and interpreted as transient expression of gusA. Photographs were taken with the digital camera of the stereomicroscope (LEICA E24HD). The transient expression of gusA was measured by counting the number of explants with at least one blue foci. The frequency of transient expression of GUS was calculated as the number of explants with at least one blue foci over the total number of explants (expressed as percentage).
Stable transformation: To assess the stability of the transformation binary and bifunctional vector pBindrd29A was used. This vector contains the selective marker nptII, which provides resistance to kanamycin and a fusion of genes containing regions of trehalose-6-phosphate synthase (tps1) and trehalose-6-phosphatase synthase (tps2) from Saccharomyces cerevisiae (Miranda et al., 2007) under the stress inducible promoter rd29A of Arabidopsis thaliana (Yamaguchi-Shinozaki & Shinozaki, 1994). This vector was added to the Agrobacterium tumefaciens LBA4404-ElectroMAX® strain. The vector and the fusion of the bifunctional genes (rd29A::tps-tpp) had a size of 16.02 and 2.9 kb, respectively (Santamaría et al., 2009). Bacterial preparation was performed as indicated previously. Transformation of common bean was carried out during two, eight, ten, 12, and 17 days of precultivation, and three and four days co-culture (Table 1).
After the co-cultivation period, bacteria were eliminated with the selected treatment of previous experiments. Explants were then cultured to RM added with 500 mg/L of timentin and 50 mg/L of kanamycin under the conditions previously described. Sub-cultures were made every three - four weeks for more than 120 days. The number of shoots surviving the selection process since the first sub-culture, was recorded. The transformation frequency of the selective gene was calculated as the number of kanamycin-resistant explants over the total number of explants.
PCR amplification of nptII and TPS1 genes: Genomic DNA was isolated from leaf tissues from four-five month old regenerated bean plants according to the extraction kit protocol MACHERY-NAGEL.
The extraction of DNA from pBindrd29A was performed according to the manual for plasmid purification (INVITROGEN). The primers used for PCR amplification correspond to the TPS1 sequence forward (5´ GTGGCAGAGGAGCTTGTTGAG 3´), and reverse (5´ GGTACTCACATACAGAC 3´), which amplify a fragment of approximately 1600 bp. The final composition of the reaction mixture was: 2.5 µl buffer 10 X, 1.0 µL Nucleotide Mix 10mM, 2 µl MgCl2 50 mM, 1 µL Forward primer (stock 10 picomol/µl) and 1 µL reverse primer (stock 10 picomol/µL), 1 µl plasmid DNA and 0.2 µl taq. The reaction conditions were: 94 °C for 5 min (1x), 30 amplification cycles (94 °C for 1 min, 50 °C for 1 min, 72 °C for 2 min), and a final extension step at 72 °C for 10 min.
The primers for the nptII gene were: forward 5´-GAGGCTATTCGGCTATGACTG and reverse 5’-TCGACAAGACCGGCTTCCATC (Aragão et al., 1996), which amplify a 410 bp fragment. The final composition of the reaction mixture was: 10 µl Master Mix 10mM, 0.25 µl forward primer (stock 10 µM/µl) and 0.25 µl reverse primer (stock 10 picomol/µl), 1 µl DNA (1:10). The reaction conditions were 94 °C for 5 min (1x), 35 amplification cycles (35 cycles of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 1 min) and a final step at 72 °C for 5 min and 4 °C.
PCR products were separated by electrophoresis on 1 % (w/v) agarose gels under 100 V for 50 min and stained with Loading Dye 6X (added with 0.6 % of Gel Red) (8 µl of PCR product was mixed with 2 µl of Loading Dye 6X 2µl molecular marker weight, 2 µl Gel Red. Gene Ruler 1 Kb Plus DNA Ladder, ready to use).
Presence of A. tumefaciens in transformed plants: The amplification by PCR of VirE2 gene fragment of A. tumefaciens was examined using the primers: forward (5’-TGCCCACCAAGGCGGAATT-3’) and reverse (5’-CTTTGCCGACCCATCGA-3’) which amplifies a fragment of 895 bp. The volume of the reaction mixture was of 25 ml containing 2.5 µl buffer 10X, 50 mM MgCl2, 10 mM dNTPS, 0.1 µl Taq polimerase, 1 µl sense primer, 1 µl antisense primer and 1 µl plant DNA. The reaction conditions were 94 °C for 30 seconds, 30 cycles of temperature of denaturing 94ºC for 5 min, primer annealing 55ºC for 5 min, elongation 72ºC for 2 min and 72 °C for 10 min and 4 °C (10 min). The amplified fragments were separed in 1 % (w/v) agarose gel to 100 V by 40 min.
All the experiments were undertaken under completely randomized designs and the statistical analysis was performed using one-way ANOVA. Means (shoots per explants, height of shoots, roots per regenerants and lenght of roots) were separated by the Duncan’s test at P < 0.05. The program SPSS Statistics version 23 was used (IBM Corp., 2015).
Results
A. tumefaciens elimination after the co-culture: Pre-cultured and co-cultured explants with LBA4404-ElectroMAX® Agrobacterium showed a 100 % bacterial suppression regardless the concentrations of cefotaxime and timentin. However, the same antibiotics treatments did not completely eliminate bacteria in non-pre-cultured explants (Table 2).
For the elimination of A. tumefaciens the most efficient and compatible with further development was the use of 500 mg/L timentin and also in the RM did not affect the in vitro response of the common bean (data not shown).
Table 2 Recidivism of LBA4404-ElectroMAX® A. tumefaciens in explants of P. vulgaris L. with or without pre-cultural times and co-cultivated during four days
| Treatments | % of Bacterial Recidivism | |
| Without pre-culture | With pre-culture | |
| Control (with neither co-culture nor antibiotics) | 0 | 0 |
| Control (with co-culture and RM without antibiotics) | 92 | 100 |
| RM+500 mg/L timentin + 500 mg/L cefotaxime | 67 | 0 |
| RM+500 mg/L timentin | 92 | 0 |
| RM+ 750 mg/L timentin | 92 | 0 |
Observations were taken after six days of starting the treatment.
Assessment of the kanamycin as selective agent: Among the assessed Kn concentrations, 25 mg/L was the only concentration that did not caused complete mortality of the explants after six weeks in culture, while concentrations of around (or higher than) 50 mg/L reached a 100 % mortality. Hence, the following experiments used a minimum lethal dose of 50 mg/L Kn to select the bean cells which were possibly transformed by the nptII gene.
Transient transformation experiments: Non-pre-cultured EAs inoculated with 0.5 OD600nm and co-cultured for three and five days showed β-glucuronidase activity in 45 % and 1.4 % of total explants, respectively (Fig. 1A). In the case of EAs co-cultured for four days, no β-glucuronidase activity was detected. Only 0.5 % of the explants pre-cultured for two days and co-cultured for four days showed gusA activity (Fig. 1 C). Furthermore, non-co-cultured explants showed an absence of endogenous GUS activity (Fig. 1B and Fig. 1D).
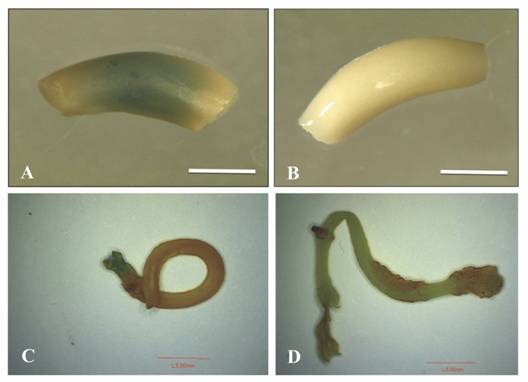
Fig. 1 GusA activity in P. vulgaris L. explants co-cultivated with A. tumefaciens LBA4404 pCAMBIA1301 with 0.5 OD600nm. A and B: non pre-cultured. C and D: pre-cultured for two days. A. co-cultivated EA for five days and B. non-infected EA (negative control), C. co-cultivated EA for four days and D. non-infected EA. Scale bars A and B= 2 mm, C and D= 5 mm.
Stable genetic transformation: Regenerants from 50 mg/L kanamycin treatment, were obtained from explants co-cultured with pBindrd29A (Table 1). These regenerants were selected 120 days after the first sub-culture (Table 3). A higher number of regenerants in the presence of the selective medium was observed in those cases under a long-term pre-culture (equal or higher than eight days). Regenerants from explants pre-cultured for 12 days and co-cultured for three days showed a higher survival rate (Table 3). Explants with a short-term pre-culture (two days) showed a low transformation frequency for the selective agent (Table 3). Fig. 2A indicates the presence of A. tumefaciens cells. In contrast, kanamycin-based selected plants did not show this amplification and the presence of the nptII gene was detected (Fig. 2B).
Table 3 Morphologic traits and frequency of regenerants of beans co-cultured with LBA4404-ElectroMAX® pBindrd29A and selected with 50 mg/L kanamycin after more than 120 days
| Experiment | Pre-culture period (days) | Co-culture period (days) | Selection period (days) | Selected npttII regenerants ( %) | Shoots per explants (Average) | height of shoots cm (Average) | Explants with roots (%) | Roots per regenerant (Average) | Length of roots cm (Average) |
| 4 | 2 | 4 | 120 | 0.5 | 2.50 ± 2.12 | 4.25 ± 3.88 | 0 | 0 ± 0 | 0 ± 0 |
| 5 | 8 | 4 | 133 | 16 | 0.10 ± 0.31 | 0.10 ± 0.31 | 10 | 0.20 ± 0.63 | 0.30 ± 0.94 |
| 6 | 10 | 4 | 133 | 19 | 1.04 ± 1.19 | 0.79 ± 1.09 | 21.73 | 0.46 ± 1.27 | 1.13 ± 3.45 |
| 7 | 12 | 3 | 148 | 36 | 0.91 ± 0.70 | 1.00 ±1.20 | 27.27 | 0.69 ± 1.54 | 1.19 ± 3.32 |
| 8 | 17 | 4 | 133 | 8 | 0 ± 0 | 0 ± 0 | 0 | 0 ± 0 | 0 ± 0 |
No significant differences between means (P < 0.05).
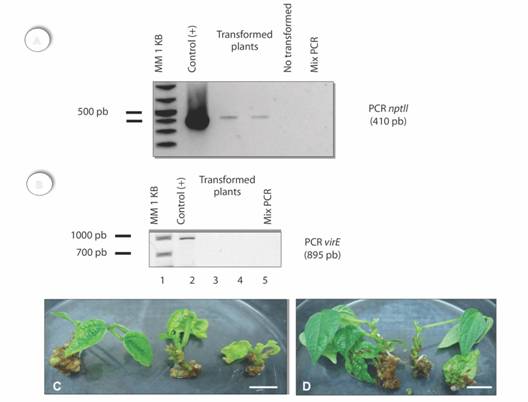
Fig. 2 Plants or common bean transformed with the nptII gene (120 days of culture). A. Amplification of a 410 bp fragment corresponding to the expected size of the selective gene. B. Absence fragment virE in transformed tissues, line 1: DNA ladder (1 Kb), line 2: Agrobacterium, Lines 3-4: regenerated and selected plants (third experiment) and line 5: load control. C. Regenerated and kanamycin-resistant plants. D. Non-transformed plants. Scale C and D bars = 1 cm.
PCR assessment of possible transformation: Plants pre-cultured and co-cultured for two and four days respectively, showed an amplification fragment of a 410 bp which corresponded to the expected size for the selective gene (Fig. 2A). Kanamycin-based selected plants did not show this amplification. Regenerants with kanamycin resistance were assessed by PCR to determine the presence of the nptII gene (Fig. 2 B). These results indicate a transformation efficiency of 0.5 % for the nptII gene in plants of common bean.
Discussion
The main limitation/constrain for obtaining transformed bean plants is the lack of an adequate genetic transformation system due to recalcitrance to the in vitro regeneration and the low Agrobacterium-mediated transformation rates.
Bacteria elimination after co-culture was also very difficult as the only way to genetically transform beans by the time this work started was by biolistics (Faria et al., 2006). Results indicated that 500 mg/L timentin eliminated the LBA4404 ElectroMax strain of Agrobacterium tumefaciens after the co-culture, which is in accordance to recent reports (Mukeshimana, Ma, Walworth, Song, & Kelly, 2013).
Bean EAs co-cultured for three days under 0.5 OD600nm and incubated at 25 ºC showed β-glucuronidase activity. This suggests a transient transformation of 45 %, which is similar to that recently reported by Collado et al. (2016), (46 %) using the C58C1RifR strain and a higher co-culture time (five days). This transformation efficiency increased (80 %) with the EHA101 strain under the same conditions (Collado et al., 2016). These results evidence the lack of endogenous gusA activity in the assessed bean explants. However, this is not the case for other species (Solís-Ramos, González-Estrada, Andrade-Torres, Godoy-Hernández, & Castaño de la Serna, 2010). The gusA reporter gene is useful for experiments related to the transformation of certain species and is suitable for histochemical staining and localization of transformed cells.
In this work, the regeneration or “shoots proliferation” from EAs was consistent with previous reports that concluded this type of explant is regenerable but not optimum for the Agrobacterium-mediated genetic transformation and subsequent shoots production (Delgado-Sánchez et al., 2006; Arellano, Fuentes, Castillo-España, & Hernández, 2009; Quintero-Jiménez, Espinosa-Huerta, Acosta-Gallegos, Guzmán-Maldonado, & Mora-Avilés, 2010; Mukeshimana et al., 2013; Solís-Ramos et al., 2015).
Optimum bacteria concentration is a relevant parameter for a transformation protocol (Collado et al., 2015). An OD600nm lower than 0.5 will not allow for genetic transformation and higher concentrations kill the explants. According to previous results (data not shown), the bacteria (LBA4404-ElectroMAX®) were used at an OD600nm of 0.5. Previous reports used similar conditions (Mukeshimana et al., 2013; Collado et al., 2015; Collado et al., 2016).
The OD600nm was combined with short-term co-cultures (three - four days). Three-days co-cultures favored the insertion of the marker gene (GUS or nptII), which agrees with the results reported by (Amugune et al., 2011). On the other hand, previous studies indicate long-term co-cultures of five days (Collado et al., 2016), six days (Collado et al., 2015), and eight days (Espinosa-Huerta et al., 2013; Mukeshimana et al., 2013) increase the temporary expression of the transgene (Zambre et al., 2003; Mukeshimana et al., 2013) especially in embryonic axes (Mukeshimana et al., 2013). The presence of BAP in the co-culture and regeneration media favored transformation and survival of the explants. Other studies report similar results (Zhang et al., 1997; Amugune et al., 2011).
Unlike other species, bean did not show natural resistance to kanamycin (Solís-Ramos et al., 2009) as 50 mg/L allowed for selecting the bean regenerants for a four -month time period. In this study, the selection was applied after the first sub-culture (four weeks) to allow transformed cells to proliferate and develop a group of cells capable to survive in middle of a mass of death cells derived from a lethal dose of antibiotic (Zambre et al., 2005). According to Amugune et al. (2011), it should be possible to recover Agrobacterium-transformed plants after bean explants had been exposed to the selection medium during three to four weeks (one month). Among all the shoots selected during more than 120 days, only four of them showed an amplification corresponding to the expected size of the ntpII gene. These results evidence that the type of the explant (embryonic axes) used in the experiment is not suitable for the regeneration of transformed plants (Mukeshimana et al., 2013), as the obtained shoots are from multicellular origin, which may inhibit the strict selection of transgenic shoots, and allows a high number of “leaks” (non-transgenic plants that survive the selection) and chimeras (Angenon & Thu, 2011). Therefore, an important contribution to enhance bean is the establishment of a regeneration system via somatic embryogenesis, or a regeneration system derived from the meristem-free tissue that minimize the production of chimeric transformants (Mukeshimana et al., 2013).
In this study, the transformation frequency obtained for the reporter and selective gene was 0.5 %, which is slightly higher to that reported for the species using biolistic 0.3 % (Aragão et al., 1996; Faria et al., 2006; Rech et al., 2008). Recently, higher transformation efficiencies (2.8 and 0.75 %) have been reported using hypervirulent strains of A. tumefaciens (EHA105 or EHA101). This affects the transfer capacity of T-DNA as chromosomal and virulence genes may differ from one strain to another (Collado et al., 2015, 2016). In the case of the TPS1 gene, the fragment was amplified in transformed explants and the controls explants, and it was not possible make a conclusion on this gene.
So far, few studies report the Agrobacterium-assisted genetic transformation for bean and no plants have been obtained, due to the lack of a regeneration protocol (Angenon & Thu, 2011; Mukeshimana et al., 2013). However, Collado et al. (2015, 2016) could recently transform beans with genetic markers (bar, nptII y uidA) producing chimeric regenerants through direct and indirect organogenesis from epicotyl. However, the established protocol did not rendered stable genetic transformation (Collado et al., 2016).
In this study, explants of common bean co-cultivated with LBA4404-ElectroMAX® from A. tumefaciens, were obtained. These showed β-glucuronidase activity indicating a temporary genetic transformation (gusA gene) of 45 % efficiency. Bean regenerants with kanamycin were selected during three to five weeks for stable transformation. However, this selection was not strict because the regenerants from multicellular origin generated chimeric transformants, leading to leaks of non-transgenic materials. Stable transformation for the nptII gene was obtained at 0.5 % efficiency, which agrees with the results related to the temporary transformation for gusA using the same protocol. The established protocol for genetic transformation of common bean has two additional advantages with respect to previous reports: (1) it allows for obtaining transformed regenerants and (2) the genetic transformation was stable for the selective gene.
Ethical statement: authors declare that they all agree with this publication and made significant contributions; that there is no conflict of interest of any kind; and that we followed all pertinent ethical and legal procedures and requirements. All financial sources are fully and clearly stated in the acknowledgements section. A signed document has been filed in the journal archives.

