










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.62 n.4 San José Oct./Dec. 2014
A Costa Rican family affected with Charcot-Marie-Tooth disease due to the myelin protein zero (MPZ) p.Thr124Met mutation shares the Belgian haplotype
Una familia costarricense afectada por la enfermedad de Charcot-Marie-Tooth, debido a la mutación p.Thr124Met en la proteína mielina cero (MPZ) compartida con el haplotipo belga
Una familia costarricense afectada por la enfermedad de Charcot-Marie-Tooth, debido a la mutación p.Thr124Met en la proteína mielina cero (MPZ) compartida con el haplotipo belga
Alejandro Leal1*, Corinna Berghoff2*, Martin Berghoff3*, Melissa Rojas-Araya4*, Carolina Ortiz5*, Dieter Heuss6*, Gerardo Del Valle7* & Bernd Rautenstrauss8*
Abstract
The p.Thr124Met mutation in the myelin protein zero (MPZ) causes the Charcot-Marie-Tooth disease type 2J, a peripheral neuropathy with additional symptoms as pupillary alterations and deafness. It was observed in several families around the world originating e. g. from Germany, Belgium, Japan, Italy and North America. Here we report Central American patients originating from a family in Costa Rica carrying this mutation. Clinical, electrophysiological and molecular analysis of patients and controls were performed, including gene and linked markers´ sequencing. Carriers share almost the entire haplotype with two non related Belgian CMT patients. As a result of the haplotype analysis, based on ten markers (seven SNPs, two microsatellites and an intronic polyA stretch), the founder effect hypothesis for this allele migration is suggestive. Rev. Biol. Trop. 62 (4): 1285-1293. Epub 2014 December 01.
Key words: Charcot-Marie-Tooth, peripheral neuropathy, Myelin protein zero, mutation, Costa Rica.
Resumen
La mutación p.thr124Met en la proteína mielina cero (MPZ) causa la enfermedad de Charcot-Marie-Tooth tipo 2J, una neuropatía periférica con síntomas adicionales como alteraciones pupilares y sordera. Se ha observado en varias familias alrededor del mundo, originarias de Alemania, Bélgica, Japón, Italia y Norteamérica, entre otras. Aquí reportamos a pacientes centroamericanos provenientes de Costa Rica que acarrean esta mutación. Se realizaron análisis clínico, electrofisiológico y molecular de pacientes y controles, incluyendo secuenciación del gen y de marcadores ligados a éste. Estos pacientes comparten casi por completo el haplotipo con dos pacientes belgas no emparentados. Como resultado del análisis de los haplotipos, basado en diez marcadores (siete SNPs, dos microsatélites y un elemento poli-A intrónico), se sugiere que se ha dado un efecto fundador en la migración de este alelo.
Palabras clave: Charcot-Marie-Tooth, neuropatía periférica, Proteína Mielina Cero, mutación, Costa Rica.
Hereditary motor and sensory neuropathy (HMSN), also called Charcot-Marie-Tooth disease (CMT), is a clinically and genetically heterogeneous group of inherited peripheral neuropathies. It is characterized by progressive muscular weakness and atrophy of the distal muscles (Lupski, 1998). By means of electrophysiological and morphological studies, CMT can be divided into two major forms: a predominantly demyelinating (HMSN type I) and an axonal form (HMSN type II) (Dyck, Chance, Lebo, & Carney, 1993).
MPZ had been initially related with a hereditary motor and sensorial neuropathy type I (HMSN I), presenting myelin disturbances resulting in reduced nerve conduction velocity (NCV<38m/s) (Hayasaka et al., 1993a). However, families with normal NCV values (HMSN II) have also been associated with MPZ (Senderek et al., 2000). The mutation Thr124Met in the third exon of the protein was found to be responsible for CMT in families with a clinically distinct phenotype, including axonal involvement, frequently deafness, Argyll Robertson-like pupils and dysphagia (Chapon, Latour, Diraison, Schaeffer, & Vandenberghe, 1999; De Jonghe et al., 1999). This mutation has been found in several CMT families. When the polymorphic markers close to the gene and an intronic polymorphism were analyzed it was associated with the same haplotype as two Belgian families. Due to this, it has been suggested that a common ancestor was the founder of this particular mutation (De Jonghe et al., 1999). Nevertheless, Senderek et al. (2000) reported a family with an identical mutation embedded in a distinct haplotype, thus suggesting the existence of a hot-spot in this codon. Not only Belgian (De Jonghe et al., 1999) families with the mutation p.Thr124Met have been reported, also French (Chapon et al., 1999), German (Senderek et al., 2000; Hanemann et al., 2001), Italian (Schiavon et al., 1998), North American (Baloh, Jen, Kim, & Baloh, 2004) and Japanese (Yoshihara et al., 2000; Kurihara et al., 2002; Numakura, Lin, Ikegami, Guldberg, & Hayasaka, 2002; Kurihara, 2003).
Here we report a Costa Rican family with CMT affected members due to the p.Thr124Met mutation in MPZ. Costa Rican family CR-C displayed autosomal dominant HMNS, clinically an intermediate form with pupillary involvement. When this phenotype was considered, a suggestion was made to test the MPZ of these families. The haplotype was studied in order to observe whether the hypothesis of a founder effect, relating the European ancestors of family CR-C with the Belgian or German families, could be supported. The Costa Rican population has a genetically major European (predominantly Spanish) component (Morera, Barrantes, & Marin-Rojas, 2003). We show evidence that the p.Thr124Met mutation in the Costa Rican family CR-C is embedded in a haplotype closely related to the Belgian patients, supporting the migration hypothesis from Europe to Central America.
Materials and methods
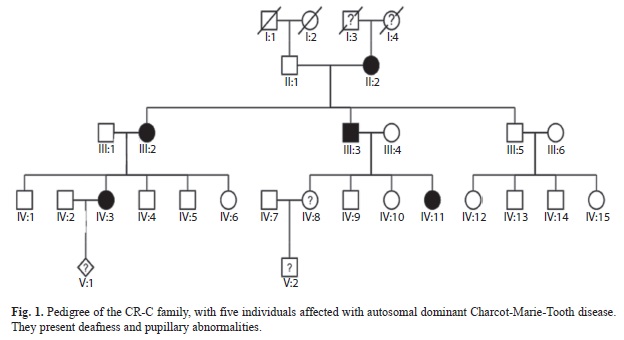
A four-generation pedigree was reconstructed based on a family originating from the Central Valley of Costa Rica, Central America (Fig. 1). Family members underwent genetic and clinical analyses, after informed consent was obtain. The study was approved by the University of Costa Rica. The clinical examination consisted of standardized tests of strength and muscular sensibility (Berghoff et al., 2004). In addition DNA from the family members was obtained from peripheral blood (from individuals II:1, II:2, III:2, III:5, IV:1, IV:3, IV:4, IV:5, IV:6, IV:8, IV:9, IV:10, IV:11, IV:12 and IV:13), via the phenol/chloroform isolation method. Standard clinical electrophysiological examinations were performed to three patients (II:2, III:2 and III:3) in the median, ulnar, peroneal and tibial nerves according to Berghoff et al. (2004).
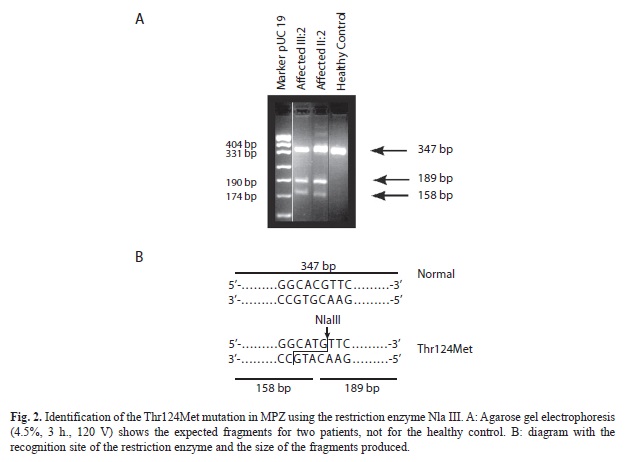
Since the Thr124Met mutation in exon 3 of MPZ alters a NlaIII restriction site the PCR product of MPZ exon 3 of all family members was digested by the restriction enzyme NlaIII (5´‘-CATG-3´). This mutation introduces a different recognition site (CACG>CATG). The primers were MPZex3-P4 (5‘-tcattagggtcctctcacatgc-3‘) and MPZex3-M21 (5´-gcctgaataaaggtccttaggc-3´) (Nelis et al., 1994). After the amplification, agarose gel electrophoresis (4.5%, 3 h., 120 V) was performed.
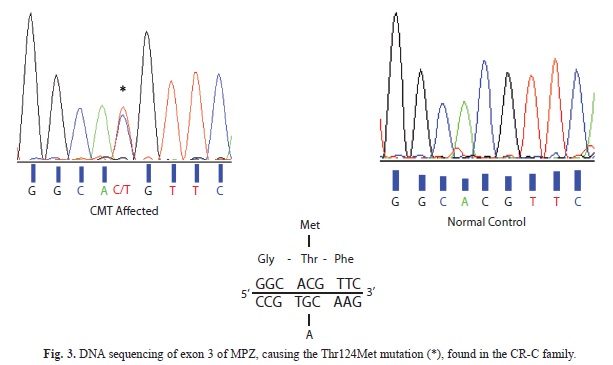
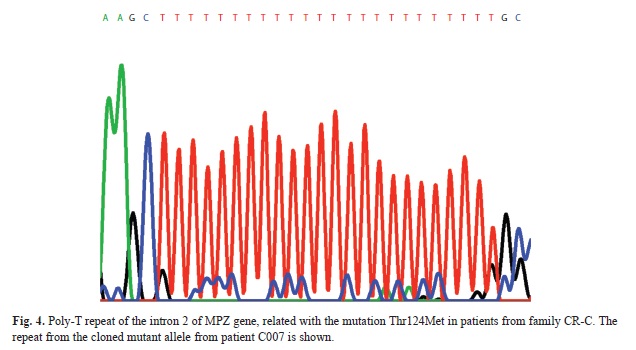
MPZ exon 3 was sequenced in both directions using an ABI 3130 DNA Sequencer (Applied Biosystems) and analyzed using Sequencing Analysis 5.2 (Applied Biosytems). In addition primer PT-F (5´-cttcctctgtatcccttactg-3´) and the reverse primer for exon 3 were used for amplifying an intronic polymorphism (poly-T repeat), to find out which alleles were linked to the mutation allele. The PCR profile used was: 94°C 20 s., 65°C to 55°C (-1°C/cycle during 10 cycles) 20 s., 68°C 45 s. and then 25 cycles at 55°C as annealing temperature. PCR products were cloned in E. coli in order to split the wild type and the mutated allele of the dT-repeat. The products were inserted in a pCR-4 Blunt-TOPO vector and TOP10 cells (Invitrogen) and transformed following the manufacturer’s instructions. Colonies were picked, and sequenced in both directions using the same primers. DNA samples from patients from families Antw-3 and Antw-5 from Belgium (De Jonghe et al., 1999) were also tested.
In addition, we examined the neighboring single nucleotide polymorphisms (SNP): rs6685495, rs6674383 using primers F: 5´-GCCCTGCATTGAGGATGTAG-3´, R: 5´-GGGACAAGGGTATGGGAAG-3´, rs12731474, rs16832793 using primers F: 5´-TGTCCTCACCTTCCTCTTGG-3´, R: 5´-GACAGTTGGGGTGGGAGTAG-3´, rs7551761, rs12065184 and rs7554208 using primers F: 5´-TCAACCATTGCCCTTCTC-AG-3´, R: 5´-TGCAATGAGTGAGCCATA-GG-3´. All seven SNPs are located in intronic sites in MPZ. The PCR profile was a “touchdown” program, starting at 94°C 20 sec., 70°C until 60°C (-1°C/cycle, during 10 cycles) 20 sec., 72°C 40 sec., and then 25 cycles at 60°C. Genotypes were obtained by sequencing in both directions using the respective primers and BigDye Terminator version 3.0 (Applied Biosystems) with an ABI 310DNA machine (Applied Biosystems) and analyzed with the SEQUENCE ANALYZER (Applied Biosystems) and SEQMAN II (DNAstar) softwares.
Results
Clinical features: The patients showed a distal symmetric motor and sensory neuropathy with additional pupillary abnormality. Mean age at onset was the fourth decade (range 19 to 68 years). Three patients (II-2, III-2, III-3, Fig. 1) had CMT diagnosis; two younger members of the family had minor symptoms (V1 and V2, Fig. 1). The oldest affected family members had distal muscle weakness, atrophy of hands and feet and progressive deafness. Furthermore, these patients showed distal sensory deficits including disturbed touch and pain perception, as well as an impaired sense of vibration and position. Spontaneous pain sensation in the legs was a predominant finding in two subjects. Three individuals complained of minor pain in the lower extremities. Three subjects reported muscle cramps in the legs. Diminished or absent deep tendon reflexes in the lower extremities were present in five subjects. All family members showed impaired pupillary reaction to light (contraction slowed). The electrophysiological data reflect axonal and demyelinating polyneuropathy with decreased amplitudes of muscle action potential, slowed nerve conduction velocity of the motor and sensory nerves and prolonged distal motor latency.
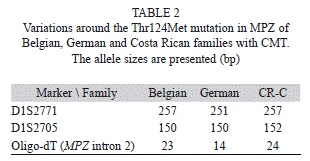
Molecular analysis: Since the particular phenotype of the patients in this family (axonally pronounced neuropathy, deafness and pupillary abnormalities) had similarities with CMT2J (OMIM 607736) patients, a digestion of the MPZ-exon 3 PCR product with the restriction enzyme Nla III was performed, resulting in two bands in an agarose electrophoresis for the patients, but only one in healthy controls (Fig. 2). The mutation was further confirmed by sequencing of exon 3 of MPZ (Fig. 3). The heterozygous mutation was observed in all affected individuals in codon 124 (C®T). The healthy controls carried the wild type sequence. Taking into account the molecular data, there was complete agreement between genotype and phenotype. Penetrance seems to be complete in this family. The only person that could develop the disease is V:1 but is still young. To investigate the origin of these variation microsatellites, SNP markers and an intronic oligo-dT repeat (Fig. 4) were used for the haplotype reconstruction, both for the Costa Rican and Belgian affected individuals. The haplotypes related with the disease in the Costa Rican and Belgian individuals are shown in table 1. Markers D1S2771, RS6674383, RS16832793, RS12731474, RS7551761 and RS7554208 were identical in affected members of both countries. In the last two markers, Belgian individuals were heterozygous, but for allele C, Costa Rican patients were homozygous. For marker RS12065184, the two Belgian individuals do not share the same genotype. Belgian individual B-003 –like the Costa Rican patients- present the allele A, and B005 the allele C. Finally, genotype of marker D1S2705 was 152 bp for the Costa Rican patients, and 150 bp for the Belgian ones, and for the oligo-dT marker, an allele of 24 repeats was present in the Costa Ricans and an allele of 23 repeats in the Belgians.
Discussion
The clinical signs of patients of the CR-C family and other families with the Thr124Met in MPZ exon 3 mutation are very similar, this suggests a constant phenotype in the mutation carriers. Most of the neuropathy patients affected by MPZ mutations represent a demyelinating hereditary motor and sensory neuropathy type I (HMSN I, CMT1B), whereas in this case the electrophysiological data reflect an intermediate type. Deafness and pupillary alteration are additional symptoms associated with this mutation. Although these three characteristics are present in CMT disease, they can also be caused by other mutations (Leal et al., 2003).
The manifestation of progressive sensorineural hearing loss and abnormal pupillary reaction may indicate that the mutation in MPZ results in an abnormality of cranial nerves in combination with and abnormality in other peripheral nerves (Seeman et al., 2004). Starr et al. (2003) found that both the sural and proximal auditory nerves showed a loss of large myelinated fibers, consistent with incomplete re-myelination; and the few distal auditory nerve fibers found in the cochlea also had a beaded appearance, consistent with incomplete re-myelination.
Clinical findings for patients of peripheral neuropathy, such as pupillary abnormalities and deafness, were associated with MPZ mutations earlier. De Jonghe et al. (1999) reported seven such families with an age of onset in the fourth and fifth decade, rapidly progressive muscle weakness and severe sensory abnormalities. Many patients initially complained of shooting pain in the legs.
In previous studies MPZ mutations have been shown to cause demyelinating neuropathy with the phenotypic variability of CMT1B (Hayasaka et al., 1993c; Gabreëls-Festen et al., 1996), Dejerine-Sottas Neuropathy (Hayasaka et al., 1993b; Rautenstrauß, Nelis, Grehl, Van Broeckhoven, & Pfeiffer, 1994; Warner et al., 1996) and congenital hypomyelination (Warner et al., 1996). As in this study, not only demyelination, but also axonal degeneration is associated with MPZ mutations (Marrosu et al., 1998; Senderek et al., 2000; Young et al., 2001). It has been suggested that axonal degeneration is most likely secondary to myelin dysfunction, possibly due to impaired axoglial interactions (Marrosu et al., 1998). Experimental studies in MPZ-deficient mice showed axonal degeneration (Giese, Martini, Lemke, Soriano, & Schachner, 1992; Frei et al., 1999) which agrees with these observations in humans. In contrast to homozygous MPZ-/- deficient mice, heterozygous MPZ+/- mutants are less severely affected. It is particularly noticeable that they do not have any signs of axonal degeneration (Martini, Zielasek, Toyka, Giese, & Schachner, 1995; Carenini et al., 2001).
Furthermore, these features are not unique in MPZ mutations since similar observations have been made in CMT related mutations in the peripheral myelin protein PMP22 gene and in neuropathy associated with gammopathy (Schröder & Himmelmann, 1992).
As mentioned, the Thr124Met mutation has been found to be responsible for CMT in families from Italy (Schiavon et al., 1998), Belgium (De Jonghe et al., 1999), France (Chapon et al., 1999; Stojkovic et al., 2003), Japan (Yoshihara et al., 2000; Misu et al., 2000), Spain (Gallardo et al., 2009), Germany (Senderek et al., 2000), and Costa Rica (this study). The haplotype analysis of the Costa Rican family showed similarity between those of Costa Rica and Belgium: both present a 257 bp-allele in D1S2771, whereas for the marker D1S2705, the difference between the allele related with the mutation in the CR-C family (152 bp) and the allele related in the Belgian family (150 bp) is only 2 bases, which could be explained by a single mutation. Also the difference between 23 oligo-dT nucleotides in the Belgian family and 24 nucleotides in the Costa Rican family could be explained by the addition of one nucleotide, or may be a sequencing artifact due to restrictions of the Sanger method. These two small changes in the markers can be explained considering the estimated disjunction time between the common European ancestors of family CR-C and the Belgian family. SNP markers are concordant in the Costa Rican and Belgian affected individual B-003. Considering the normal mutation rate in a single nucleotide in an unexpressed sequence is 2.5 x 10-8 mutations per nucleotide site or 175 mutations per diploid genome per generation (Nachman & Crowell, 2000), most SNPs-variants in the region should be identical if these families have a common origin. This is what is observed in the Costa Rican and Belgian families. The haplotypes similarity indicates a founder effect behind the disease in these families. Interestingly, the SNP (RS12065184) is different in the two Belgian patients. However, the rest of the haplotype is identical, thus the hypothesis of an additional mutation at this place is probable.
On the other hand, Costa Rican and Belgian haplotypes differ with the German ones (Senderek et al., 2000) (Table 2). This fact suggests that the mutation locus has also properties of a hot spot, prone to variation. As conclusion, this mutation can be explained by a founder effect and as well the existence of an error prone site at codon 124 of MPZ. Further genealogical studies may elucidate the known historical Flemish influence in Latin America.
Acknowledgments
We thank the family members who collaborated in this study for giving their informed consent, personal information and blood samples. We also thank C. Van Broeckhoven and J. Senderek for DNAs from Belgian and German affected individuals. Support from the Vicerrectoría de Investigación of the University of Costa Rica is gratefully acknowledged (project 111-A4-334). BR was supported by the DFG.
References
Baloh, R. H., Jen, J. C., Kim, C., & Baloh, R.W. (2004). Chronic cough due to Thr124Met mutation in the peripheral myelin protein zero (MPZ gene). Neurology, 62(10), 1905-1906. [ Links ]
Berghoff, C., Berghoff, M., Leal, A., Morera, B., Barrantes, R., Reis, A., Neundoerfer, B., Rautenstrauss, B., Del Valle, G., & Heuss, D. (2004). Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (CMT2B2) that maps to chromosome 19q13.3. Neuromuscular Disorders, 14, 301-306. [ Links ]
Carenini, S., Maurer, M., Werner, A., Blazyca, H., Toyka, K. V., Schmid, C. D., Raivich, G., & Martini, R. (2001). The role of macrophages in demyelinating peripheral nervous system of mice heterozygously deficient in p0. Journal of Cellular Biology, 152, 301-308. [ Links ]
Chapon, F., Latour, P., Diraison, P., Schaeffer, S., & Vandenberghe, A. (1999). Axonal phenotype of Charcot-Marie Tooth disease associated with a mutation in the myelin protein zero gene. Journal of Neurology, Neurosurgery and Psychiatry, 66, 779-782. [ Links ]
De Jonghe, P., Timmerman, V., Ceuterick, C., Nelis, E., De Vriendt, E., Löfgren, A., Vercruyssen, A., Verellen, C., Van Maldergem, L., Martin, J., & Van Broeckhoven, C. (1999). The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain, 122, 281-290. [ Links ]
Dyck, P., Chance, P., Lebo, R., & Carney, J. (1993). Hereditary motor and sensory neuropathies. In P. Dyck, P. Thomas, J. Griffin, P. Low, & J. Poduslo (Eds.), Peripheral neuropathies (pp. 1094-1136). Philadelphia: Saunders. [ Links ]
Frei, R., Motzing, S., Kinkelin, I., Schachner, M., Koltzenburg, M., & Martini, R. (1999). Loss of distal axons and sensory Merkel cells and features indicative of muscle denervation in hindlimbs of P0-deficient mice. Journal of Neuroscience, 19, 6058-6067. [ Links ]
Gabreëls-Festen, A. A., Hoogendijk, J. E., Meijerink, P. H., Gabreëls, F. J., Bolhuis, P. A, van Beersum, S., Kulkens, T., Nelis, E., Jennekens, F. G., de Visser, M., van Engelen, B. G., Van Broeckhoven, C., & Mariman, E. C. (1996). Two divergent types of nerve pathology in patients with different P0 mutations in Charcot-Marie-Tooth disease. Neurology, 47, 761-765. [ Links ]
Gallardo, E., García, A., Ramón, C., Maraví, E., Infante, J., Gastón, I., Alonso, A., Combarros, O., Jonghe, P. D., & Berciano, J. (2009). Charcot-Marie–Tooth disease type 2J with MPZ Thr124Met mutation: clinico-electrophysiological and MRI study of a family. Journal of Neurology, 256, 2061-2071. [ Links ]
Giese, K. P., Martini, R., Lemke, G., Soriano, P., & Schachner, M. (1992). Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell, 71, 565-576. [ Links ]
Hanemann, C. O., Gabreëls-Festen, A. A., & De Jonghe, P. (2001). Axon damage in CMT due to mutation in myelin protein P0. Neuromuscular Disorders, 11(8), 753-756. [ Links ]
Hayasaka, K., Himoro, M., Sato, W., Takada, G., Uyemura, K., Shimizu, N., Bird, T. D., Conneally, P. M., & Chance, P. F. (1993a). Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P(0) gene. Nature Genet, 5, 31-34. [ Links ]
Hayasaka, K., Himoro, M., Sawaishi, Y., Nanao, K., Takahashi, T., Takada, G., Nicholson, G. A., Ouvrier, R. A., & Tachi, N. (1993b). De novo mutation of the myelin Po gene in Dejerine-Sottas disease (hereditary motor and sensory neuropathy type III). Nature Genetics, 5, 266-268. [ Links ]
Hayasaka, K., Himoro, M., Wang, Y., Takata, M., Minoshima, S., Shimizu, N., Miura, M., Uyemura, K., & Takada, G. (1993c). Structure and chromosomal localization of the gene encoding the human myelin protein zero (MPZ). Genomics, 17, 755-758. [ Links ]
Kurihara, S., Adachi, Y., Wada, K., Adachi, A., Ohama, E., & Nakashima, K. (2003). Axonal and demyelinating forms of the MPZ Thr124Met mutation. Acta Neurologica Scandinavica, 108, 157-160. [ Links ]
Kurihara, S., Adachi, Y., Wada, K., Awaki, E., Harada, H., & Nakashima, K. (2002). An epidemiological genetic study of Charcot-Marie-Tooth disease in Western Japan. Neuroepidemiology, 21(5), 246-250. [ Links ]
Leal, A., Berghoff, C., Berghoff, M., Del Valle, G., Contreras, C., Montoya, O., Hernández, E., Barrantes, R., Schoetzer-Schrehardt, U., Neundoerfer, B., Reis, A., Rautenstrauss, B., & Heuss, D. (2003). Charcot-Marie-Tooth disease: a novel Tyr145Ser mutation in the myelin protein zero (MPZ, P0) gene causes different phenotypes in homozygous and heterozygous carriers within one family. Neurogenetics, 4, 191-197. [ Links ]
Lupski, J. (1998). Molecular genetics of peripheral neuropathies. In J. Martin (Ed.), Scientific American Molecular Neurology (pp. 239-256). New York: Scientific American Library. [ Links ]
Marrosu, M. G., Vaccargiu, S., Marrosu, G., Vannelli, A., Cianchetti, C., & Muntoni, F. (1998). Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene. Neurology, 50, 1397-1401. [ Links ]
Martini, R., Zielasek, J., Toyka, K. V., Giese, K. P., & Schachner, M. (1995). Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nature Genetics, 11, 281-286. [ Links ]
Morera, B., Barrantes, R., & Marin-Rojas, R. (2003). Gene admixture in the Costa Rican population. Annals of Human Genetics, 67, 71-80. [ Links ]
Nelis, E., Timmerman, V., De Jonghe, P., Vandenberghe, A., Pham-Dinh, D., Dautigny, A., Martin, J. J., & Van Broeckhoven, C. (1994). Rapid screening of myelin genes in CMT1 patients by SSCP analysis: identification of new mutations and polymorphisms in the P0 gene. Human Genetics, 94(6), 653-657. [ Links ]
Misu, K., Yoshihara, T., Shikama, Y., Awaki, E., Yamamoto, M., Hattori, N., Hirayama, M., Takegami, T., Nakashima, K., & Sobue, G. (2000). An axonal form of Charcot–Marie–Tooth disease showing distinctive features in association with mutations in the peripheral myelin protein zero gene (Thr124Met or Asp75Val). Journal of Neurology, Neurosurgery and Psychiatry, 69, 806-811. [ Links ]
Nachman, M. W. & Crowell, S. L. (2000). Estimate of the mutation rate per nucleotide in humans. Genetics, 156, 297-304. [ Links ]
Numakura, C., Lin, C., Ikegami, T., Guldberg, P., & Hayasaka, K. (2002). Molecular analysis in Japanese patients with Charcot-Marie-Tooth disease: DGGE analysis for PMP22, MPZ, and Cx32/GJB1 mutations. Human Mutation, 20(5), 392-398. [ Links ]
Rautenstrauß, B., Nelis, E., Grehl, H., Pfeiffer, R. A., & Van Broeckhoven, C. (1994) Identification of a de novo insertional mutation in P0 in a patient with Déjerine-Sottas phenotype. Human Molecular Genetics, 3(9), 1701-1702. [ Links ]
Schiavon, F., Rampazzo, A., Merlini, L., Angelini, C., & Mostacciuolo, M. L. (1998). Mutations of the same sequence of the myelin P0 gene causing two different phenotypes. Human Mutation, 11, S217-S219. [ Links ]
Schröder, J. M. & Himmelmann, F. (1992). Fine structural evaluation of altered Schmidt-Lanterman incisures in human sural nerve biopsies. Acta Neuropatholica (Berlin), 83, 120-133. [ Links ]
Seeman, P., Mazanec, R., Huehne, K., Suslikova, P., Keller, O., & Rautenstrauss, B. (2004). Hearing loss as the first feature of leta-onset axonal CMT disease due to a novel P0 mutation. Neurology, 63, 733-735. [ Links ]
Senderek, J., Hermanns, B., Lehmann, U., Bergmann, C., Marx, G., Kabus, C., Timmerman, V., Stoltenburg-Didinger, G., & Schroder, J. M. (2000). Charcot-Marie-Tooth neuropathy type 2 and P0 point mutations: two novel amino acid substitutions (Asp61Gly; Tyr119Cys) and a possible “hotspot” on Thr124Met. Brain Pathology, 10, 235-248. [ Links ]
Starr, A., Michalewski, H. J., Zeng, F. G., Fujikawa-Brooks, S., Linthicum, F., Kim, C. S., Winnier, D., & Keats, B. (2003). Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145→àSer). Brain, 126, 1-16. [ Links ]
Stojkovic, T., de Seze, J., Dubourg, O., Arne-Bes, M. C., Tardieu, S., Hache, J. C., & Vermersch, P. (2003). Autonomic and respiratory dysfunction in Charcot–Marie–Tooth disease due to Thr124Met mutation in the myelin protein zero gene. Clinical Neurophysiology, 114, 1609-1614. [ Links ]
Warner, L. E., Hilz, M. J., Appel, H., Killian, J. M., Kolodny, E. H., Carpenter, S., Watters, G. V., Wheeler, C., Witt, D., Bodell, A., Nelis, E., Van Broeckhoven, C., & Lupski, J. R. (1996). Clinical phenotypes of different mpz (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron, 17, 451-460. [ Links ]
Yoshihara, T., Yamamoto, M., Doyu, M., Mis, K. I., Hattori, N., Hasegawa, Y., Mokuno, K., Mitsuma, T., & Sobue, G. (2000). Mutations in the peripheral myelin protein zero and connexin32 genes detected by non-isotopic RNase cleavage assay and their phenotypes in Japanese patients with Charcot-Marie-Tooth disease. Human Mutation, 16, 177-178. [ Links ]
Young, P., Grote, K., Kuhlenbaumer, G., Debus, O., Kurlemann, H., Halfter, H., Funke, H., Ringelstein, E. B., & Stogbauer, F. (2001). Mutation analysis in Chariot-Marie Tooth disease type 1: point mutations in the MPZ gene and the GJB1 gene cause comparable phenotypic heterogeneity. Journal of Neurology, 248, 410-415. [ Links ]
Berghoff, C., Berghoff, M., Leal, A., Morera, B., Barrantes, R., Reis, A., Neundoerfer, B., Rautenstrauss, B., Del Valle, G., & Heuss, D. (2004). Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (CMT2B2) that maps to chromosome 19q13.3. Neuromuscular Disorders, 14, 301-306. [ Links ]
Carenini, S., Maurer, M., Werner, A., Blazyca, H., Toyka, K. V., Schmid, C. D., Raivich, G., & Martini, R. (2001). The role of macrophages in demyelinating peripheral nervous system of mice heterozygously deficient in p0. Journal of Cellular Biology, 152, 301-308. [ Links ]
Chapon, F., Latour, P., Diraison, P., Schaeffer, S., & Vandenberghe, A. (1999). Axonal phenotype of Charcot-Marie Tooth disease associated with a mutation in the myelin protein zero gene. Journal of Neurology, Neurosurgery and Psychiatry, 66, 779-782. [ Links ]
De Jonghe, P., Timmerman, V., Ceuterick, C., Nelis, E., De Vriendt, E., Löfgren, A., Vercruyssen, A., Verellen, C., Van Maldergem, L., Martin, J., & Van Broeckhoven, C. (1999). The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain, 122, 281-290. [ Links ]
Dyck, P., Chance, P., Lebo, R., & Carney, J. (1993). Hereditary motor and sensory neuropathies. In P. Dyck, P. Thomas, J. Griffin, P. Low, & J. Poduslo (Eds.), Peripheral neuropathies (pp. 1094-1136). Philadelphia: Saunders. [ Links ]
Frei, R., Motzing, S., Kinkelin, I., Schachner, M., Koltzenburg, M., & Martini, R. (1999). Loss of distal axons and sensory Merkel cells and features indicative of muscle denervation in hindlimbs of P0-deficient mice. Journal of Neuroscience, 19, 6058-6067. [ Links ]
Gabreëls-Festen, A. A., Hoogendijk, J. E., Meijerink, P. H., Gabreëls, F. J., Bolhuis, P. A, van Beersum, S., Kulkens, T., Nelis, E., Jennekens, F. G., de Visser, M., van Engelen, B. G., Van Broeckhoven, C., & Mariman, E. C. (1996). Two divergent types of nerve pathology in patients with different P0 mutations in Charcot-Marie-Tooth disease. Neurology, 47, 761-765. [ Links ]
Gallardo, E., García, A., Ramón, C., Maraví, E., Infante, J., Gastón, I., Alonso, A., Combarros, O., Jonghe, P. D., & Berciano, J. (2009). Charcot-Marie–Tooth disease type 2J with MPZ Thr124Met mutation: clinico-electrophysiological and MRI study of a family. Journal of Neurology, 256, 2061-2071. [ Links ]
Giese, K. P., Martini, R., Lemke, G., Soriano, P., & Schachner, M. (1992). Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell, 71, 565-576. [ Links ]
Hanemann, C. O., Gabreëls-Festen, A. A., & De Jonghe, P. (2001). Axon damage in CMT due to mutation in myelin protein P0. Neuromuscular Disorders, 11(8), 753-756. [ Links ]
Hayasaka, K., Himoro, M., Sato, W., Takada, G., Uyemura, K., Shimizu, N., Bird, T. D., Conneally, P. M., & Chance, P. F. (1993a). Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P(0) gene. Nature Genet, 5, 31-34. [ Links ]
Hayasaka, K., Himoro, M., Sawaishi, Y., Nanao, K., Takahashi, T., Takada, G., Nicholson, G. A., Ouvrier, R. A., & Tachi, N. (1993b). De novo mutation of the myelin Po gene in Dejerine-Sottas disease (hereditary motor and sensory neuropathy type III). Nature Genetics, 5, 266-268. [ Links ]
Hayasaka, K., Himoro, M., Wang, Y., Takata, M., Minoshima, S., Shimizu, N., Miura, M., Uyemura, K., & Takada, G. (1993c). Structure and chromosomal localization of the gene encoding the human myelin protein zero (MPZ). Genomics, 17, 755-758. [ Links ]
Kurihara, S., Adachi, Y., Wada, K., Adachi, A., Ohama, E., & Nakashima, K. (2003). Axonal and demyelinating forms of the MPZ Thr124Met mutation. Acta Neurologica Scandinavica, 108, 157-160. [ Links ]
Kurihara, S., Adachi, Y., Wada, K., Awaki, E., Harada, H., & Nakashima, K. (2002). An epidemiological genetic study of Charcot-Marie-Tooth disease in Western Japan. Neuroepidemiology, 21(5), 246-250. [ Links ]
Leal, A., Berghoff, C., Berghoff, M., Del Valle, G., Contreras, C., Montoya, O., Hernández, E., Barrantes, R., Schoetzer-Schrehardt, U., Neundoerfer, B., Reis, A., Rautenstrauss, B., & Heuss, D. (2003). Charcot-Marie-Tooth disease: a novel Tyr145Ser mutation in the myelin protein zero (MPZ, P0) gene causes different phenotypes in homozygous and heterozygous carriers within one family. Neurogenetics, 4, 191-197. [ Links ]
Lupski, J. (1998). Molecular genetics of peripheral neuropathies. In J. Martin (Ed.), Scientific American Molecular Neurology (pp. 239-256). New York: Scientific American Library. [ Links ]
Marrosu, M. G., Vaccargiu, S., Marrosu, G., Vannelli, A., Cianchetti, C., & Muntoni, F. (1998). Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene. Neurology, 50, 1397-1401. [ Links ]
Martini, R., Zielasek, J., Toyka, K. V., Giese, K. P., & Schachner, M. (1995). Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nature Genetics, 11, 281-286. [ Links ]
Morera, B., Barrantes, R., & Marin-Rojas, R. (2003). Gene admixture in the Costa Rican population. Annals of Human Genetics, 67, 71-80. [ Links ]
Nelis, E., Timmerman, V., De Jonghe, P., Vandenberghe, A., Pham-Dinh, D., Dautigny, A., Martin, J. J., & Van Broeckhoven, C. (1994). Rapid screening of myelin genes in CMT1 patients by SSCP analysis: identification of new mutations and polymorphisms in the P0 gene. Human Genetics, 94(6), 653-657. [ Links ]
Misu, K., Yoshihara, T., Shikama, Y., Awaki, E., Yamamoto, M., Hattori, N., Hirayama, M., Takegami, T., Nakashima, K., & Sobue, G. (2000). An axonal form of Charcot–Marie–Tooth disease showing distinctive features in association with mutations in the peripheral myelin protein zero gene (Thr124Met or Asp75Val). Journal of Neurology, Neurosurgery and Psychiatry, 69, 806-811. [ Links ]
Nachman, M. W. & Crowell, S. L. (2000). Estimate of the mutation rate per nucleotide in humans. Genetics, 156, 297-304. [ Links ]
Numakura, C., Lin, C., Ikegami, T., Guldberg, P., & Hayasaka, K. (2002). Molecular analysis in Japanese patients with Charcot-Marie-Tooth disease: DGGE analysis for PMP22, MPZ, and Cx32/GJB1 mutations. Human Mutation, 20(5), 392-398. [ Links ]
Rautenstrauß, B., Nelis, E., Grehl, H., Pfeiffer, R. A., & Van Broeckhoven, C. (1994) Identification of a de novo insertional mutation in P0 in a patient with Déjerine-Sottas phenotype. Human Molecular Genetics, 3(9), 1701-1702. [ Links ]
Schiavon, F., Rampazzo, A., Merlini, L., Angelini, C., & Mostacciuolo, M. L. (1998). Mutations of the same sequence of the myelin P0 gene causing two different phenotypes. Human Mutation, 11, S217-S219. [ Links ]
Schröder, J. M. & Himmelmann, F. (1992). Fine structural evaluation of altered Schmidt-Lanterman incisures in human sural nerve biopsies. Acta Neuropatholica (Berlin), 83, 120-133. [ Links ]
Seeman, P., Mazanec, R., Huehne, K., Suslikova, P., Keller, O., & Rautenstrauss, B. (2004). Hearing loss as the first feature of leta-onset axonal CMT disease due to a novel P0 mutation. Neurology, 63, 733-735. [ Links ]
Senderek, J., Hermanns, B., Lehmann, U., Bergmann, C., Marx, G., Kabus, C., Timmerman, V., Stoltenburg-Didinger, G., & Schroder, J. M. (2000). Charcot-Marie-Tooth neuropathy type 2 and P0 point mutations: two novel amino acid substitutions (Asp61Gly; Tyr119Cys) and a possible “hotspot” on Thr124Met. Brain Pathology, 10, 235-248. [ Links ]
Starr, A., Michalewski, H. J., Zeng, F. G., Fujikawa-Brooks, S., Linthicum, F., Kim, C. S., Winnier, D., & Keats, B. (2003). Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145→àSer). Brain, 126, 1-16. [ Links ]
Stojkovic, T., de Seze, J., Dubourg, O., Arne-Bes, M. C., Tardieu, S., Hache, J. C., & Vermersch, P. (2003). Autonomic and respiratory dysfunction in Charcot–Marie–Tooth disease due to Thr124Met mutation in the myelin protein zero gene. Clinical Neurophysiology, 114, 1609-1614. [ Links ]
Warner, L. E., Hilz, M. J., Appel, H., Killian, J. M., Kolodny, E. H., Carpenter, S., Watters, G. V., Wheeler, C., Witt, D., Bodell, A., Nelis, E., Van Broeckhoven, C., & Lupski, J. R. (1996). Clinical phenotypes of different mpz (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron, 17, 451-460. [ Links ]
Yoshihara, T., Yamamoto, M., Doyu, M., Mis, K. I., Hattori, N., Hasegawa, Y., Mokuno, K., Mitsuma, T., & Sobue, G. (2000). Mutations in the peripheral myelin protein zero and connexin32 genes detected by non-isotopic RNase cleavage assay and their phenotypes in Japanese patients with Charcot-Marie-Tooth disease. Human Mutation, 16, 177-178. [ Links ]
Young, P., Grote, K., Kuhlenbaumer, G., Debus, O., Kurlemann, H., Halfter, H., Funke, H., Ringelstein, E. B., & Stogbauer, F. (2001). Mutation analysis in Chariot-Marie Tooth disease type 1: point mutations in the MPZ gene and the GJB1 gene cause comparable phenotypic heterogeneity. Journal of Neurology, 248, 410-415. [ Links ]
1. Section of Genetics and Biotechnology, School of Biology and Neuroscience Research Center, University of Costa Rica, San José, Costa Rica. Institute of Human Genetics, University of Erlangen-Nuremberg, Erlangen, Germany; alejandro.leal@ucr.ac.cr
2. Practice of Neurology, Giessen, Germany; corinna.berghoff@t-online.de
3. Department of Neurology, University of Giessen, Giessen, Germany; martin.berghoff@neuro.med.uni-giessen.de
4. Section of Genetics and Biotechnology, School of Biology and Neuroscience Research Center, University of Costa Rica, San José, Costa Rica; memaroar@yahoo.com
5. Section of Genetics and Biotechnology, School of Biology and Neuroscience Research Center, University of Costa Rica, San José, Costa Rica; carolinaortizco@gmail.com
6. Department of Neurology, University of Erlangen-Nuremberg, Erlangen, Germany; dieter.heuss@uk-erlangen.de
7. Laboratory of Neurophysiology, Neurolab, San José, Costa Rica; neurolab22@yahoo.com
8. Medizinisch Genetisches Zentrum, Munich, Germany. Institute of Human Genetics, University of Erlangen-Nuremberg, Erlangen, Germany; rautenstrauss@mgz-muenchen.de
Received 02-II-2014. Corrected 10-VII-2014. Accepted 13-VIII-2014.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}