










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.62 n.2 San José Apr./Jun. 2014
Microsatellites loci reveal heterozygosis and population structure in vampire bats (Desmodus rotundus) (Chiroptera: Phyllostomidae) of Mexico
Microsatélites revelan heterocigosidad y estructura poblacional de murciélagos vampiro (Desmodus rotundus) (Chiroptera: Phyllostomidae) de México
Microsatélites revelan heterocigosidad y estructura poblacional de murciélagos vampiro (Desmodus rotundus) (Chiroptera: Phyllostomidae) de México
*Dirección para correspondencia:
Abstract
A limited number of studies have focused on the population genetic structure of vampire bats (Desmodus rotundus) in America. This medium-sized bat is distributed in tropical areas of the continent with high prevalence in forested livestock areas. The aim of this work was to characterize the vampire population structure and their genetic differentiation. For this, we followed standard methods by which live vampires (caught by mist-netting) and preserved material from scientific collections, were obtained for a total of 15 different locations, ranging from Chihuahua (North) to Quintana Roo (Southeast). Tissue samples were obtained from both live and collected animals, and the genetic differentiation, within and among localities, was assessed by the use of seven microsatellite loci. Our results showed that all loci were polymorphic and no private alleles were detected. High levels of heterozygosis were detected when the proportion of alleles in each locus were compared. Pairwise FST and RST detected significant genetic differentiation among individuals from different localities. Our population structure results indicate the presence of eleven clusters, with a high percentage of assigned individuals to some specific collecting site. Rev. Biol. Trop. 62 (2): 659-669. Epub 2014 June 01.
Key words: heterozygosity, México, microsatellite, population structure, vampire bats.
Resumen
Muy pocos trabajos se han enfocado en el estudio genético de las poblaciones de vampiro (Desmodus rotundus) en América. Este murciélago de tamaño mediano se encuentra distribuido en las áreas tropicales de América, con una gran prevalencia en zonas de ganadería. La recolecta de tejidos se realizó mediante redes de niebla y en con ejemplares de colecciones, estas dan un total de 15 localidades. Mediante el uso de siete microsatellites, nosotros estudiamos la diferenciación genética dentro y entre localidades muestreadas, estas fueron desde Chihuahua en el norte, hasta Quintana Roo en el sureste. Todos los loci fueron polimórficos y no se encontraron alelos privados. Se encontraron altos niveles de heterócigos cuando se compararon la proporción de alelos en cada locus. Comparaciones pareadas de FST y RST mostraron una diferenciación genética entre los individuos de las diferentes localidades. Los resultados de estructura genética indican la presencia de once clusters, con un alto porcentaje de asignación de los individuos a las localidades en donde fueron recolectados.
Palabras clave: estructura poblacional, heterocigosidad, México, microsatélite, vampiros.
Phyllostomid bats are often abundant and widely distributed in the tropical and subtropical areas of America, seemingly due the evolutionary origin of the group and its local adaptations to appropriate tropical habitats (Wetterer, Rockman, & Simmons, 2000). Some phyllostomid species are more tolerant to human disturbance and extreme conditions of fragmented habitats, these species are adapted to a semi-natural matrix (a mix of original habitat with different levels of human induced disturbance), and may function as a measure of habitat integrity (Medellín, Equihua, & Amin, 2000). Several authors evaluated the ecological role of phyllostomid bats as indicators of habitat disturbance (Johns, Wilson, & Pine, 1985; Fenton et al., 1992; Medellín, et al., 2000), showing how some phyllostomids are linked to human disturbances. For instance, the Jamaican fruit-eating bat (Artibeus jamaicensis) is a common component of the urban fauna in tropical Middle America. Similarly, the high abundance of vampire bats (Desmodus rotundus) has been linked to the growth of livestock production (Medellín et al., 2000).
The genetic structure of wild animals reveals information about population size, dispersal, reproductive success, mating system, relatedness, among much other potential information (Kerth, Safi, & König, 2002). Locally abundant populations are expected to maintain high levels of gene flow, augmented genetic diversity, and to have high genetic divergence when compared to geographically distant populations. Genetic structure is defined as genetically differentiated populations due to physical barriers to migration combined with the dispersal ability of the species. Species with limited dispersal abilities show more population structure than species with greater dispersal abilities (Storz, Bhat, & Kunz, 2001; Chen, et al., 2010). High bat vagility is expected when competition for local resources is high, allowing individuals to commute large distances (Chen et al., 2010). However, vampire bats (D. rotundus) are not limited by scarce food availability due to the introduction of cattle in tropical areas (Lee, Papes, & Van Den Bussche, 2012). Landscape structure may play an important role in the genetic diversity and local gene flow of species, but has been probed that D. rotundus accomplishment high densities on patchy habitats especially influenced by cattle density and remains of original vegetation (Lee et al., 2012). Thus, differences in genetic structure among populations of vampire bats may depend upon the geographic scale of the study, and on local factors that allow/restrict gene flow of the species.
The vampire bat is a widely distributed species, ranging from Northern Mexico to Northern Chile and occupying the entire Amazon basin (Greenhall, Joermann, & Schmidt, 1983). Vampire bats live in colonies that generally consist of hundreds of individuals; females form long-lasting associations by sharing food with their roostmates (Wilkinson, 1985a). Additionally, self-grooming and social grooming has been reported in colonies of vampire bats occupying hollow trees, and this behavior correlates with the ectoparasite load of each individual but does not have a genetic component (Wilkinson, 1986). Some colonies are composed by dominant males and fewer unrelated females, resembling a social structure of polygamy (Wilkinson, 1985b). Vampire bats from different geographic mtDNA clades with the biogeographic pattern revealing strong population structure suggesting the possibility of cryptic species (Martins, Ditchfield, Meyer, & Morgante, 2007). Martins, Templeton, Pavan, Kohlbach, and Morgante (2009), examined vampire bats samples from Central America and Brazil by using mitochondrial and nuclear markers. Their results revealed geographical structure with a historical scenario with mtDNA but no phylogeographic structure with nuclear markers and suggested that these contrasting patterns are compatible with complete isolation in Pleistocene refuges. Although these previous works examined genetic diversity in vampire bats populations in different regions, they did not study genetic structure and genetic diversity in the Northern most range of its distribution.
We studied the vampire bat population genetics to evaluate genetic diversity, genetic structure and genetic divergence among different populations along its distributional range in Mexico. Our objective was to show that genetic diversity and structure of vampire bat populations are linked to the collecting site and limited migrations, with a null impact at larger scales because vampire bats do not perform broad migratory movements. We analyzed the genetic diversity of vampire bat populations and evaluated our results. We hypothesized genetic population structure with a high percent of assigned individuals to some specific areas. We show that vampire bats can benefit with human disturbance in the context of genetic diversity by presenting our data in different quantitative ways.
Materials and methods
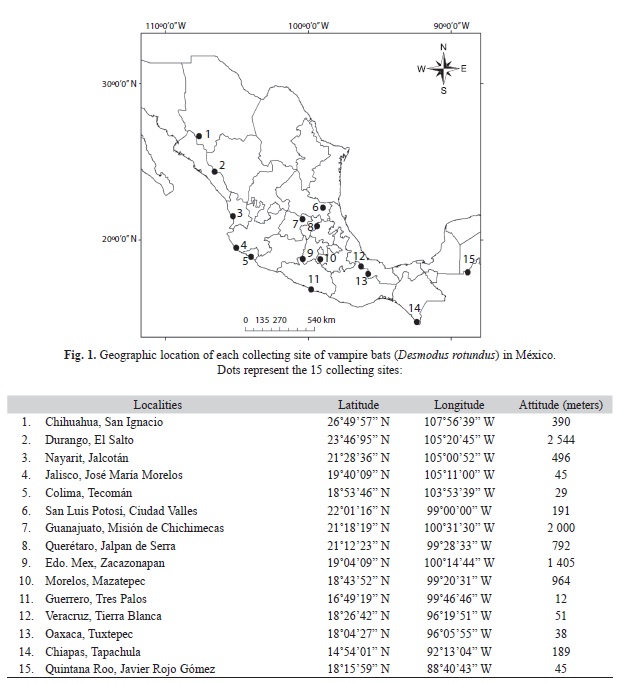
Sampling collection and DNA extraction: Wing samples were obtained from 15 different locations in Mexico between 2005 and 2010. For some locations at least 10 individuals were captured, but for some others, tissues were donated by established collections (Fig. 1). For this, we mist-net different ecosystems in the selected localities, for a sampling effort of 4 to 5 nights per locality, and of five hours (from 19:00 to 24:00hrs). Specifically, vampire bats were collected, using mist nets set among different habitats of the same locality (e. g. tropical deciduous forest (n=7), or forested cattle area (n=8). Mist nets were placed three consecutive nights at each locality. Captured individuals were considered to be adults when the wing epiphyses were completely ossified, and to be juveniles when the joints were cartilaginous. Standard morphological measurements were taken to corroborate this classification. A spring scale (exact to 0.1mm) was used for body mass; and a mechanical caliper (exact to 0.1mm) was used for total length and forearm length measurements. Wing samples were collected and stored in ethanol (96%) at -70°C; after this procedure bats were marked with gentian violet and released. Whole genomic DNA was extracted following instructions of the Qiagen protocol (Blood and Tissue Kit, Cat No. 69504). Quality of DNA was assessed by 1% agarose gel electrophoresis in combination with molecular weight standards.
Microsatellite genotyping: We used seven dinucleotide microsatellite loci designed specifically for D. rotundus (Piaggio, Johnston & Perkins, 2008). PCR conditions consisted of an initial denaturation at 95°C for 10 min, followed by 30 cycles at 94°C for 30s, Ta for 45s (see original for each primer), and 72°C for 45s, and a final extension of 72°C for 10 min. All reactions were performed in a Perkin Elmer 9700 Thermalcycler. Amplification of microsatellites was carried out in a 15μL volume containing 30ng of DNA, 0.2μM of each primer, 0.2μM of dNTP´s, 1x Taq buffer (1.5μM of MgCl2, 10mM of Tris-HCl, 50mM of KCl), and 0.75U of Taq polymerase. Analysis was performed on an ABI Prism 3100 Genetic Analyzer. Analysis of computer generated results was executed using the GeneScan (version 2.1) software and final allele-sizing was done with the ABI Genotyper package (version 2.1).
Allelic frequencies, F statistics (Weir, 1996), RST (Rousset, 1996), Hardy-Weinberg equilibrium, and genotypic disequilibrium among all loci pairs, as well expected and observed heterozygosities were estimated using the software program GENEPOP 3.1b (Raymond & Rousset, 1995) and ARLEQUIN v 3.0 to estimate pairwise statistics (Excoffier, Smouse & Quattro, 1992). We also estimated allelic richness and private allele richness with correction for sample size through rarefaction using the software HP-RARE (Kalinowski, 2005). The software MICROCHECKER (Van Oosterhout, Hutchinson, Willis, & Shipley, 2004) was used to test for the presence of null alleles through the Brookfield method (Brookfield, 1996). Genetic structure was examined using AMOVA in ARLEQUIN V 3.0 (Excoffier et al., 1992; Excoffier, Laval, & Schneider, 2005), with 1 000 repetitions and confidence intervals based on 20 000 repetitions. We also used SAMOVA ver. 1.0, which considers spatial information, to obtain the locality of groups that maximize the cluster value and better explain the distribution of the genetic variance (Dupanloup, Schneider, & Excoffier, 2002).
To examine levels of genetic divergence, POPULATIONS Version 1.2.28 (O. Langella, Centre National de la Recherche Scientifique, Laboratoire Populations, Génétique et Evolution, Gif sur Yvette; HYPERLINK "http://www.cnrs-gif.fr/pge/bioinfo/populations" http://www.cnrs-gif.fr/pge/bioinfo/populations) was used to generate a Nei’s standard genetic distance matrix (Saitou & Nei, 1987). To determine the degree of population genetic structure, we used the program STRUCTURE Version 2 (Pritchard, Stephens, & Donnelly, 2000). This program uses Bayesian analysis to cluster individuals into subpopulations (K) with no prior information as to known populations. In this case, 100 000 MCMC repetitions following a 100 000 burn-in period was used and run ten times independently for K=1 to 10. An admixture model was used and correlated allele frequencies were assumed. The mean posterior probability was calculated for each set of ten runs of K and used to determine an optimal K.
Results
Sampling collection data: We collected 181 vampire bats from 15 different localities in Mexico. Vampire bats were more frequently captured in forested cattle than in tropical deciduous forest areas (paired t-test, t=0.72, p<0.05). Our samples of adult vampire bats showed no deviation from a 1:1 sex ratio (two tailed binomial test, females=85, males=96; p=0.89). Adults did not vary in size according to their sampled locality. Neither length of forearm or body mass differed among the 15 localities (one-way ANOVA, F=6.22, d.f.=14, p>0.05 for length of forearm, F=5.81, d.f.=14, p>0.05). Reproductive condition of males showed scrotal testes, but female pregnancy was infrequently reported.
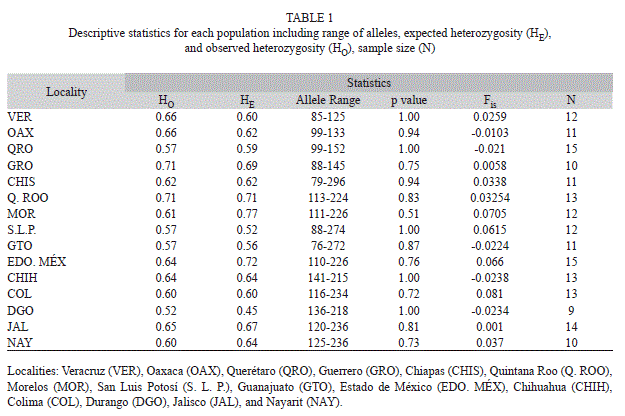
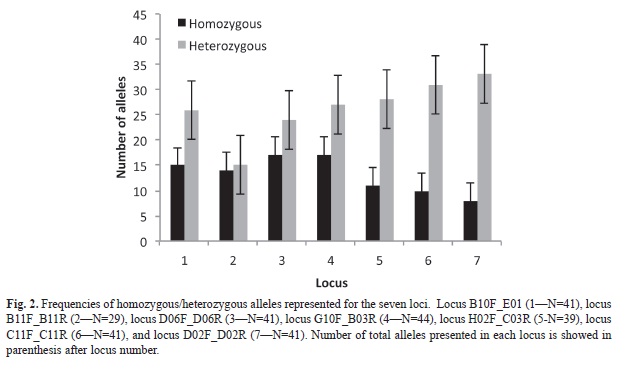
Microsatellite data results: We detected 221 alleles for the seven nuclear microsatellite loci. All loci were polymorphic (range between 23-36 alleles per locus) for all localities. No null alleles or linkage disequilibrium was detected. All localities no presented deviations from HWE (Table 1). Observed heterozygosity was similar than expected heterozygosity for all populations. For almost all loci, the number of heterozygotes was higher than homozygotes (Mann-Whitney U-test, U=4.78, p<0.05, Fig. 2). Subsequent multiple comparison showed no difference for two loci (B10F_ E01R and D06F_D06R), but a significant differences among homozygote/heterozygote relationship on the rest.
Morelos was the population with the highest allelic richness (28±0.45) and genetic diversity (0.73±0.12), followed by Estado de México and Guerrero. Durango was the population with the lowest allelic richness (14±0.73) and genetic diversity (0.12±0.07), followed by San Luis Potosí. Several private alleles were found among different populations.
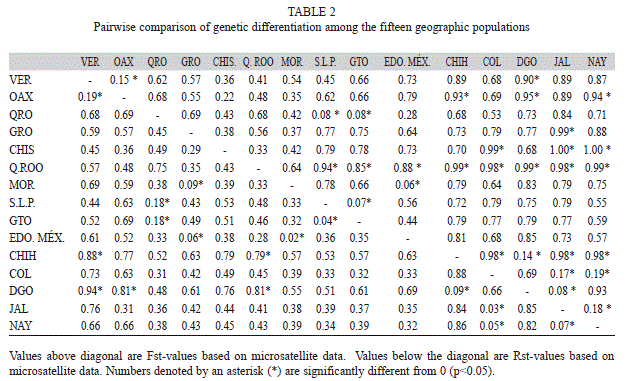
The 27% of pairwise RST comparisons were significant (RST>0; p<0.05), with the highest genetic differentiation between Chihuahua and Quintana Roo populations (1.00) and the lowest significant value between Morelos and Estado de México populations (0.06) (Table 2). Chihuahua and Durango in the North, and Quintana Roo in Southeastern, were the populations that showed the greatest genetic differentiation from the rest of the populations. FST comparisons, 15 % were significant, with the highest genetic differentiation between Durango and Veracruz (0.94), and the lowest among Jalisco and Colima (0.03).
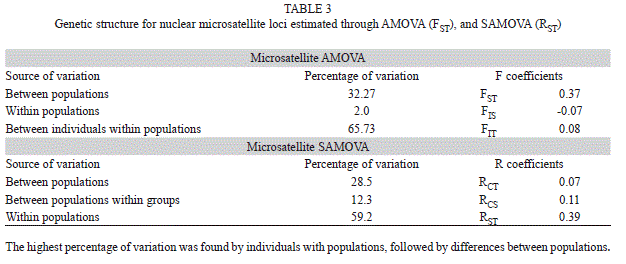
A hierarchical AMOVA was performed to detect population genetic structure. For the defined populations, 32.27% of the variation was detected among populations, 65.73% of the variation was partitioned by individuals within populations (Table 3). Fixation indexes showed genetic structure between populations (FST=0.37; p<0.05) with a low inbreeding coefficient among them (FIS=-0.07 and FIT=0.08; p>0.05). Considering SAMOVA results, the spatial distribution identified two clusters among populations: 1) between Estado de México and Morelos, and 2) between Jalisco and Nayarit. Values of SAMOVA distribution remained similar as those presented with AMOVA, with the highest value found by individuals within populations (59.2), and followed by differences among populations (28.5, Table 3).
Population structure could be detected using the Bayesian cluster approach (Evanno, Regnaut & Goudet, 2005). The STRUCTURE program suggested that the sampled D. rotundus most likely represent defined groups. Eleven population clusters were identified without any prior population information. Estado de México and Morelos were assigned to one group, whereas Nayarit+Colima+Jalisco represented other assigned cluster. Guanajuato and Querétaro formed a cluster mainly consisted by individuals of the studied population, but representatives of Jalisco are included in this group. While most of the 181 vampire bats could undoubtedly be assigned to one of the 11 clusters, some individuals exhibited genotypes that might originate outside of the cluster sampled, suggesting some degree of admixture.
Discussion
Vampire bats (D. rotundus) are restricted to tropical and subtropical areas of America. Specimens of vampire bats are relatively easy to obtain due their local abundance, wide range and feeding behavior. Information about the biology of the species is abundant and based on individuals of its distributional range (Greenhall et al., 1983). In recent decades however, additional data emerged and improved our understanding of the ecological role of this species (Aguilar-Sétien et al., 1998; Voigt & Kelm, 2006; Streicker et al., 2012). The present study represents the largest sampling of D. rotundus in México to date, and indicates that genetic diversity is high for all sampled populations, with well-defined populations related to the collected areas.
Our molecular genetic data provides insight into the population structure of D. rotundus. Genetic evidence for the existence of these clusters comes from our analyses of nuclear microsatellite data. Our analyses of the geographical distribution of microsatellite variability at seven nuclear loci further corroborate assignation with highest likelihood to inferred populations in the STRUCTURE analysis. F-statistics indicate a highly significant nuclear differentiation between few collected localities. Genetic differentiation between populations of vampire bats in America leads to high levels of sequence divergence and cryptic species in Desmodus complex (Martins et al., 2007; Pinto, 2009; Hernández-Dávila et al., 2012). Although D. rotundus is restricted to the tropical areas of the Neotropics, extensive field surveys suggest that D. rotundus remains well distributed within its fragmented range, and abundant within forested habitats with available food (Lee et al., 2012). A wide range with low vagility presumably facilitates population isolation between localities, promoting the development of significant genetic structure among populations. Our data suggest that recent gene flow among D. rotundus populations is quite limited; however, in our results some neighboring localities clustered in the assignment test. Our results warrant further research based on larger sample sizes and knowledge of dispersal rates that have been inferred in other bat species (Russell, Medellín, & McCracken, 2005).
We found significant excesses in heterozygotes as measured by one statistical comparison in populations of Desmodus rotundus in Mexico. High levels of heterozygosis in mammals has been inadequately reported (i. e. black tailed prairie dogs-Foltz & Hoogland, 1983; baboons-Huchard, 2010; domestic sheep- Smith, Hoffman, Green, & Amos, 2011), and its evidence is poorly understood. Heterozygosity excess has been appointed to several causes (Stoeckel et al., 2006): 1) May possibility the result from small reproductive populations, where only few breeders have real fitness. 2) Outbreeding may be result of selective forces of the most heterozygous individuals. 3) Could be the result of asexual reproduction, or 4) random mating behavior in large dense populations. We discarded hypothesis three because the natural biology of the species. Small reproductive populations of D. rotundus it is not possible because the usual abundance of the species (Medellín et al., 2000). The most plausible explanation for high levels of heterozygosity would be dense local residents of vampire bats with no assortative mating behavior, under some natural selection forces.
We support the idea of large, abundant, year round breeding populations of vampire bats in tropical disturbed localities of Mexico. D. rotundus is one of the common genera in order of abundance in the Neotropics and is considered an important element of the species richness of fragmented areas by its profusion (Trajano, 1996; Kraker-Castañeda & Echeverría-Tello, 2012; Lee et al., 2012). Vampire bats have a clear positive response to environmental disturbance, with an increase in abundance related to the growth in livestock areas (Medellín et al. 2000; Coutinho & Bernard, 2012; Streicker et al., 2012). Haematophagous bats are continuously exposed to virus such as rabies, and strong selective forces must be acting to preserve the prevalence of the disease in the Neotropics (Mayen, 2003; Salmón-Mulanovich et al., 2009). Overall nuclear genetic diversity in D. rotundus was higher than reported for other bat species (Rossiter, Jones, Ransome, & Barratt, 2000; Ortega, Maldonado, Fleischer, Arita, & Wilkinson, 2003; Asher, 2009). It is well-known that reproductive seasonality is absent in vampire bats because they present active reproductive conditions year-round due the food disposition, and compare with other bats that are limited in food availability (Nuñez & de Viana, 1997; Wimsatt & Trapido, 2005). Levels of genetic diversity in vampire bats could be negatively affected by its control campaigns such as dynamiting caves, shotguns, smoked and fired caves, etc. (Mickleburgh, Hutson & Racey, 2002). Survey data suggest high local flow of vampire bats in suburban and livestock areas (Langoni et al., 2008), and no-relatedness relationship among cave roost mates (Wilkinson, 1985b).
High levels of genetic divergence and population differentiation among our sampling sites may increase over time, especially due to changes from tropical original habitats to livestock areas. Our results showed that our conclusions are consistent with expected genetic diversity and population differentiation for a species that benefits from human activities. Perhaps we expected less diversity in peripheral populations (i. e. Chihuahua Eckert, Samis, & Lougheed, 2008), but this population exhibit the same level of genetic diversity like the rest of sampled populations. Increased livestock areas with remnants of original tropical forest, in addition with the existence of suitable roosting sites (i. e. caves), supports an appropriate habitat for a positive growth in vampire bats populations. Therefore, the increased livestock area cover and the mosaic of tropical forest may benefit vampire bat populations and facilitate dispersal among fragments. Future research should evaluate the sampling of populations throughout the distributional range of the species, particularly in South America.
Acknowledgments
Financial support was provided by CONACyT Ciencia Básica (156725). Some tissue samples were obtained from A. Rendón (Instituto Tecnológico de la Cuenca del Papaloapan), L. León-Paniagua (Museo de Zoología “Alfonso L. Herrera” UNAM), C. López (CIIDIRDurango, Instituto Politécnico Nacional), J. A. Guerrero (Universidad Autónoma del Estado de Morelos). First author thanks Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional by all facilities. Collecting permit was provided by SEMARNAT.
References
Aguilar-Sétien, A., Brochier, B., Tordo, N., de Paz, O., Desmettre, P., Péharpré, D., & Pastoret, P. P. (1998). Experimental rabies infection and oral vaccination in vampire bats (Desmodus rotundus). Vaccine, 16, 1122-1126. [ Links ]
Asher, C. (2009). Patterns of genetic diversity in populations of two bat species (Sturnira ludovici and Artibeus toltecus) in Cusuco National Park, Honduras. Bioscience Horizons, 2, 147-154. [ Links ]
Brookfield, J. F. Y. (1996). A simple new method for estimating null allele frequency from heterozygote deficiency. Molecular Ecology, 5, 453-455. [ Links ]
Chen, J. S., Rossiter, J., Flanders, J. R., Sun,Y., Hua, P., Miller-Butterworth, C., Liu, X., Rajan, K. E., & Zhang, S. (2010). Contrasting genetic structure in two co-distributed species of old world fruit bat. PLOS ONE 5, e13903. doi:10.1371/journal.pone.0013903 [ Links ]
Coutinho, G. C. & Bernard, E. (2012). Neotropical bats as indicators of environmental disturbance: What is the emerging message? Acta Chiropterologia, 14, 143-151. [ Links ]
Dupanloup, I., Schneider, S., & Excoffier, L. (2002). A simulated annealing approach to define the genetic structure of populations. Molecular Ecology, 11, 2571-2581. [ Links ]
Eckert, C. G., Samis, K. E., & Lougheed, S. C. (2008). Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Molecular Ecology, 17, 1170-1188. [ Links ]
Evanno, G., Regnaut, S., & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology, 14, 2611-2620. [ Links ]
Excoffier, L., Laval, G., & Schneider, S. (2005). Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 1, 47-50. [ Links ]
Excoffier, L., Smouse, P., & Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131, 479-491. [ Links ]
Fenton, M. B., Acharya, L., Audet, D., Hickey, M. B. C., Merriman, C., Obrist, M. K., Syme, D. M., & Adkins, D. (1992). Phyllostomid bats (Chiroptera: Phyllostomidae) as indicators of habitat disruption in the Neotropics. Biotropica, 24, 440-446. [ Links ]
Foltz, D. W. & Hoogland, J. L. (1983). Genetic evidence of outbreeding in the black-tailed prairie dog (Cynomys ludovicianus). Evolution, 37, 273-281. [ Links ]
Greenhall, A. M., Joermann, G., & Schmidt, U. (1983). Desmodus rotundus. Mammalian Species, 202, 1-6. [ Links ]
Hernández Dávila, A., Vargas, J. A., Martínez-Méndez, N., Lim, B. K., Engstrom, M., & Ortega, J. (2012). DNA Barcoding and genetic diversity of phyllostomid bats from Yucatan Peninsula with comparisons with Central America. Molecular Ecology Resources, 12, 590-597. [ Links ]
Huchard, E., Knaap, L. A., Wang, J., Raymon, M., & Cowlishaw, G. (2010). MHC, mate choice and heterozygote advantage in a wild social primate. Molecular Ecology, 19, 2545 2561. [ Links ]
Johns, A. D., Wilson, D. E., & Pine, R. H. (1985). Rain forst bats: an uncertain future. Bat News, 5, 4-5. [ Links ]
Kalinowski, S. T. (2005). HP-RARE 1.0: A computer program for performing rarefaction measures of allelic richness. Molecular Ecology Notes, 5, 187-189. [ Links ]
Kerth, G., Safi, K., & König, B. (2002). Mean colony relatedness is a poor predictor of colony structure and female philopatry in the communally breeding Bechstein´s bat (Myotis bechsteinii). Behavioral Ecology & Sociobiology, 52, 203-210. [ Links ]
Kraker-Castañeda, C. & Echeverría-Tello, J. L. (2012). Riqueza de especies y variabilidad trófica de murciélagos en zonas de riesgo de rabia de origen silvestre en Izabal, Guatemala. Therya, 3, 87-99. [ Links ]
Langoni, H., Souza, L. C., Zetun, C. B., Silva, T. C. C., Hoffman, J. L., & Silva, R. C. (2008). Serological survey for rabies in serum samples from vampire bats (Desmodus rotundus) in Botucatu region, SP, Brazil. Journal of Venom Animals and Toxins including Tropical Diseases 1, 651-659. [ Links ]
Lee, D. N., Papes, M., & Van Den Bussche, R. A. (2012). Present and potential future distribution of common vampire bats in the Americas and the associated risk to cattle. PLoS ONE 7, e42466.doi:10.1371/journal.pone.0042466. [ Links ]
Martins, F. M., Ditchfield, A. D., Meyer, D., & Morgante, J. S. (2007). Mitochondrial DNA phylogeography reveals marked population structure in the common vampire bat, Desmodus rotundus (Phyllostomidae). Journal of Zoological Systematics and Evolutionary Research, 45, 372-378. [ Links ]
Martins, F. M., Templeton, A. R., Pavan, A. C., Kohlbach, B. C., & Morgante, J. S. (2009). Phylogeography of the common vampire bat (Desmodus rotundus): Marked population structure, Neotropical Pleistocene vicariance and incongruence between nuclear and mtDNA markers. BMC Evolutionary Biology, 9, 294-306. [ Links ]
Mayen, F. (2003). Haematophagous bats in Brazil, their role in rabies transmission, impact on public health, livestock industry and alternatives to an indiscriminate reduction of bat population. Journal of Veterinary Medicine, B 50, 469-472. [ Links ]
Medellín, R. A., Equihua, M. & Amin, M. A. (2000). Bat diversity and abundance as indicators of disturbance in Neotropical rain forests. Conservation Biology, 14, 1666-1675. [ Links ]
Mickleburgh, S. P., Hutson, A. M., & Racey, P. A. (2002). A review of the global conservation status of bats. Oryx, 36, 18-34. [ Links ]
Nuñez, H. A. & de Viana, M. L. (1997). Estacionalidad reproductiva en el vampiro común Desmodus rotundus (Chiroptera, Phyllostomidae) en el Valle de Lerma (Salta, Argentina). Revista de Biología Tropical, 45, 1231-1235. [ Links ]
Ortega, J., Maldonado, J. E., Fleischer, R. S., Arita, H. T., & Wilkinson, G. S. (2003). Male dominance, paternity, and relatedness in the Jamaican fruit-eating bat (Artibeus jamaicensis). Molecular Ecology, 12, 2409-2415. [ Links ]
Piaggio, A. J., Johnston, J. J., & Perkins, S. L. (2008). Development of microsatellite loci for the common vampire bat, Desmodus rotundus (Chiroptera: Phyllostomidae). Molecular Ecology Resources, 8, 440-442. [ Links ]
Pinto, C. M. (2009). Genetic diversity of the common vampire bat Desmodus rotundus in Ecuador: Testing cross-Andean gene flow. (M. Sc. Thesis). Texas Tech University, Texas. [ Links ]
Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945-959. [ Links ]
Raymond, M. & Rousset, F. (1995). GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity, 86, 248-249. [ Links ]
Rossiter, S. J., Jones, G., Ransome, R. D., & Barratt, E. M. (2000). Genetic variation and population structure in the endangered greater horseshoe bat Rhinolophus ferrumequinum. Molecular Ecology, 9, 1131-1135. [ Links ]
Rousset, F. (1996). Equilibrium Values of Measures of Population Subdivision for Stepwise Mutation Processes. Genetics, 14, 1357-1362. [ Links ]
Russell, A. L., Medellín, R. A., & McCracken, G. F. (2005). Genetic variation and migration in the Mexican freetailed bat (Tadarida brasiliensis mexicana). Molecular Ecology, 14, 2207-2222. [ Links ]
Saitou, N., & Nei, M. (1987). The neighbor-joining method: A new method for reconstruction phylogenetic trees. Molecular Biology and Evolution, 4, 406-425. [ Links ]
Salmón-Mulanovich, G., Vásquez, A., Albújar, Guevara, C., Laguna-Torres, C. A., Salazar, M., Zamalloa, H., Cáceres, M., Gómez-Benavides, J., Pacheco, V., Contreras, C., Kochel, T., Niezgoda, M., Jackson, F. R., Velasco-Villa, A., Rupprecht, C. E., & Montgomery, J. M. (2009). Human rabies and rabies in vampire and nonvampire bat species, Southeastern Peru, 2007. Emergent Infective Diseases, 15, 1308-1310. [ Links ]
Smith, E. M., Hoffman, J. I., Green, L. E., & Amos, W. (2011). Preliminary association of microsatellite heterozygosity with footrot in domestic sheep. Livestock Science, 143, 293-299. [ Links ]
Stoeckel, S., Grange, J., Fernández-Manjarres, J. F., Bilger, I., Frascaria-Lacoste, N., & Mariette, S. (2006). Heterozygote excess in a self-incompatible and partially clonal forest tree species-Prunus avium L. Molecular Ecology, 15, 2109-2118. [ Links ]
Storz, J. F., Bhat, H. R., & Kunz, T. H. (2001). Genetic consequences of polygyny and social structure in an Indian fruit bat, Cynopterus sphinx. I. Inbreeding, outbreeding, and population subdivision. Evolution, 55, 1215-1223. [ Links ]
Streicker, D. G., Recuenco, S., Valderrama, W., Gomez- Benavidez, J., Vargas, I., Pacheco, V., Condori-Condori, R. E., Montgomery, J., Rupprecht, C. E., Rohani, P., & Altizer, S. (2012). Ecological and anthropogenic drivers of rabies exposure in vampire bats: implication for transmission and control. Proceedings of the Royal Society, B 279, 3384-3392. [ Links ]
Trajano, E. (1996). Movements of cave bats in Southeastern Brazil, with emphasis on the population ecology of the common vampire bat, Desmodus rotundus (Chiroptera). Biotropica, 28, 121-129. [ Links ]
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M., & Shipley, P. (2004). MICROCHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes, 4, 535-538. [ Links ]
Voigt, C. C. & Kelm, D. H. (2006). Host preference of the common vampire bat (Desmodus rotundus; Chiroptera) assessed by stable isotopes. Journal of Mammalogy, 87, 1-6. [ Links ]
Weir, B. S. (1996). Genetic data analysis II: Methods for discrete population genetic data. Sinauer, Sunderland, USA. [ Links ]
Wetterer, A. L., Rockman, M. V., & Simmons, N. B. (2000). Phylogeny of phyllostomid bats (Mammalia: Chiroptera): data from diverse morphological systems, sex chromosomes, and restriction sites. Bulletin of the American Museum of Natural History, 248, 1-200. [ Links ]
Wilkinson, G. S. (1985a). The social organization of the common vampire bat. I. Pattern and cause of association. Behavioral Ecology and Sociobiology, 17, 111-121. [ Links ]
Wilkinson, G. S. (1985b). The social organization of the common vampire bat. II. Mating system, genetic structure, and relatedness. Behavioral Ecology and Sociobiology, 17, 123 134. [ Links ]
Wilkinson, G. S. (1986). Social grooming in the common vampire bat, Desmodus rotundus. Animal Behaviour, 34, 1880-1889. [ Links ]
Wimsatt, W. A. & Trapido, H. (2005). Reproduction and the female reproductive cycle in the tropical American vampire bat, Desmodus rotundus murinus. American Journal of Anatomy, 91, 415-445. [ Links ]
* Correspondencia a:
Claudia Romero-Nava. Laboratorio de Bioconservación y Manejo, Posgrado en Ciencias Quimicobiológicas, Departamento de Zoología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Prolongación de Carpio y Plan de Ayala s/n, Col. Sto. Tomas, 11340, México, D. F.; yayita_2804@hotmail.com
Livia León-Paniagua. Museo de Zoología “Alfonso L. Herrera”, Facultad de Ciencias, Universidad Nacional Autónoma de México. Apartado Postal 70-399, México D.F., 04510; llp@ciencias.unam.mx
Jorge Ortega. Laboratorio de Bioconservación y Manejo, Posgrado en Ciencias Quimicobiológicas, Departamento de Zoología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Prolongación de Carpio y Plan de Ayala s/n, Col. Sto. Tomas, 11340, México, D. F.; artibeus2@aol.com
1. Laboratorio de Bioconservación y Manejo, Posgrado en Ciencias Quimicobiológicas, Departamento de Zoología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Prolongación de Carpio y Plan de Ayala s/n, Col. Sto. Tomas, 11340, México, D. F.; yayita_2804@hotmail.com
2. Museo de Zoología “Alfonso L. Herrera”, Facultad de Ciencias, Universidad Nacional Autónoma de México. Apartado Postal 70-399, México D.F., 04510; llp@ciencias.unam.mx
3. Laboratorio de Bioconservación y Manejo, Posgrado en Ciencias Quimicobiológicas, Departamento de Zoología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Prolongación de Carpio y Plan de Ayala s/n, Col. Sto. Tomas, 11340, México, D. F.; artibeus2@aol.com
Received 14-v-2013. Corrected 10-Xi-2013. Accepted 10-Xii-2013.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}