










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.56 n.1 San José Mar. 2008
Fernando Morales1,3, Patricia Cuenca1,2,3, Gerardo del Valle4, Melissa Vásquez1,3, Roberto Brian5, Mauricio Sittenfeld6, Keith Johnson7, Xi Lin8 & Tetsuo Ashizawa8
1 Instituto de Investigaciones en Salud, Universidad de Costa Rica, Ciudad Universitaria Rodrigo Facio, San José, Costa Rica. Tel: 207-3050. Fax: 207-5130; famorale@cariari.ucr.ac.cr
2 Escuela de Biología, Universidad de Costa Rica, San José, Costa Rica.
3 Programa de Investigación en Neurociencias, Universidad de Costa Rica, San José, Costa Rica.
4 Laboratorio de Neurofisiologia (Neurolab), Curridabat, San José, Costa Rica
5 Servicio de Neurología, Hospital Nacional de Niños, San José, Costa Rica
6 Servicio de Neurología, Hospital San Juan de Dios, San José, Costa Rica
7 Division of Molecular Genetics, Institute of Biomedical and Live Sciences, University of Glasgow, Anderson College Building, 56 Dumbarton Road, Glasgow G11 6NU, Scotland, UK
8 Department of Neurology, University of Texas Medical Branch, Galveston, Texas 77555-0144, Texas, USA
Abstract: Myotonia congenita is a muscular disease characterized by myotonia, hypertrophy, and stiffness. It is inherited as either autosomal dominant or recessive known as Thomsen and Becker diseases, respectively. Here we confirm the clinical diagnosis of a family diagnosed with a myotonic condition many years ago and report a new mutation in the CLCN1 gene. The clinical diagnosis was established using ocular, cardiac, neurological and electrophysiological tests and the molecular diagnosis was done by PCR, SSCP and sequencing of the CLCN1 gene. The proband and the other affected individuals exhibited proximal and distal muscle weakness but no hypertrophy or muscular pain was found. The myotatic reflexes were lessened and sensibility was normal. Electrical and clinical myotonia was found only in the sufferers. Slit lamp and electrocardiogram tests were normal. Two affected probands presented diminution of the sensitive conduction velocities and prolonged sensory distal latencies. The clinical spectrum for this family is in agreement with a clinical diagnosis of Becker myotonia. This was confirmed by molecular diagnosis where a new disease-causing mutation (Q412P) was found in the family and absent in 200 unaffected chromosomes. No latent myotonia was found in this family; therefore the ability to cause this subclinical sign might be intrinsic to each mutation. Implications of the structure-function-genotype relationship for this and other mutations are discussed. Adequate clinical diagnosis of a neuromuscular disorder would allow focusing the molecular studies toward the confirmation of the initial diagnosis, leading to a proper clinical management, genetic counseling and improving in the quality of life of the patients and relatives. Rev. Biol. Trop. 56 (1): 1-11. Epub 2008 March 31.
Keywords: myotonia congenita, myotonic dystrophy, Becker myotonia, chloride channelopathy, SSCP.
The term myotonia refers to a feature of the skeletal muscle mechanics, which is characterized by a lengthening in the muscle relaxation time that occurs after a voluntary or mechanical stimuli, resulting in a transitory failure to complete the antagonic movement (Morales et al. 2003). Diseases associated with this symptom are collectively termed myotonias and accordingly to their clinical features, they are classified into: 1-dystrophic myotonias and 2-non-dystrophic myotonias. Myotonic dystrophy type 1 (DM1) and 2 (DM2), the most common muscular problem in young adults, belong to the first group, whereas the sodium channelopathies and the chloride channelopathies or myotonia congenita belong to the second (reviewed in Morales and Cuenca 2004).
Myotonia congenita (MC) is a hereditary muscular disease, electrophysiologically characterized by presenting increased excitability of the muscular fiber, which is due to repetitive action potentials of the muscle membranes, which is reflected in clinical myotonia, muscular stiffness and hypertrophy (Meyer-Kleine et al. 1995, Zhang et al. 1996). The clinical phenotype depends partially on whether the disease is inherited as autosomal dominant, (termed Thomsen disease) or as an autosomal recessive generalized myotonia (termed Becker disease). However, the latter is clinically more severe and more common (Sun et al. 2001). The two disorders differ clinically by the age of onset, spreading of the myotonia, a typical transient muscular weakness only present in the recessive trait and genetically by their transmission pattern (Koch et al. 1992, Koch et al. 1993).
Onset of myotonia congenital (Thomsen and Becker disease) is early in childhood (during the first or second decade of life), but usually earlier in Thomsen disease (Nagamitsu et al. 2000, Jurkat-Rott et al. 2002, Heatwole and Moxley 2007). Muscular stiffness can affect every skeletal muscle in the body, but is ameliorated by exercise (warm-up phenomenon). It can be associated with transient weakness during quick movements lasting only seconds or as long as thirty minutes in Becker disease (Jurkat-Rott et al. 2002). Nevertheless, myotonia in MC is clinically highly variable, ranging from only EMG detectable myotonic discharges to disabling muscle stiffness at an early age (Sun et al. 2001).
The two diseases are associated with mutations in the CLCN1 gene, located in chromosome 7q35. The CLCN1 gene has 23 exons and encodes the skeletal muscle chloride channel protein (CLC-1) with 18 a-helix domains, some of these being transmembrane domains (Koch et al. 1992). CLCN1 is a voltage-gate dependent channel belonging to the CLC family of chloride channels, of which nine members have been identified thus far (Grunnet et al. 2003). This channel is a complex homodimer that conducts chloride ions over the entire physiological voltage ranges and is consequently the major mediator of chloride conductance in the skeletal muscle (Esteban et al. 1998, Zhang et al. 2000b, Jurkat-Rott et al. 2002). The intensive search for mutations that followed its discovery showed that the dominant form is very rare, since very few different families (~14 families) have been shown to present a clear dominant inheritance pattern (reviewed in Colding-Jørgensen 2005). The recessive form is more common, with a frequency of ~1:23 000. Males seem to be affected predominately over females with a ratio of 3:1 only when the typical clinical features are taken into account. However, family studies indicate that women are affected at the same frequency, although to a much lesser degree (Lehmann-Horn and Jurkat-Rott 1999). Eighty different CLCN1 mutations have been associated with MC (Colding-Jørgensen 2005). The majority of these mutations are found in the recessive form (Becker disease), whereas only a few have been described with the pure dominant form (Thomsen disease) (Pusch 2002, Colding-Jørgensen 2005). A clear distinction between dominant and recessive mutations is not always possible, since several mutants have been described in both recessive and dominant traits (Meyer-Kleine et al. 1995, Koty et al. 1996, Sun et al. 2001, Pusch 2002, Colding-Jørgensen 2005).
It was originally suggested that the chloride channel was a dimer with an unusual structure; two independent pores forming a so-called "double-barrel", with two independent fast-gating mechanism and one slow-gating mechanism (Grunnet et al. 2003). X-ray data have elucidated the structure of the chloride channel (Dutzler et al. 2002), however little is known about the structure/function relationship of Cl-channel (Jurkat-Rott et al. 2002). The "double-barrel" model proposed by this study can explain the dual inheritance of congenital myotonic mutations in a recessive or dominant manner (Grunnet et al. 2003).
The aim of our study was to establish the clinical and molecular diagnosis of a Costa Rican family that had not had an adequate clinical diagnosis since the first cases in the family appeared. Here we report clinical and molecular data from a family carrying a new mutation in the CLCN1 gene causing Becker disease and discuss the possible implications of the mutations and the function-structure-phenotype relationships in the CLCN1 channel.
Materials and methods
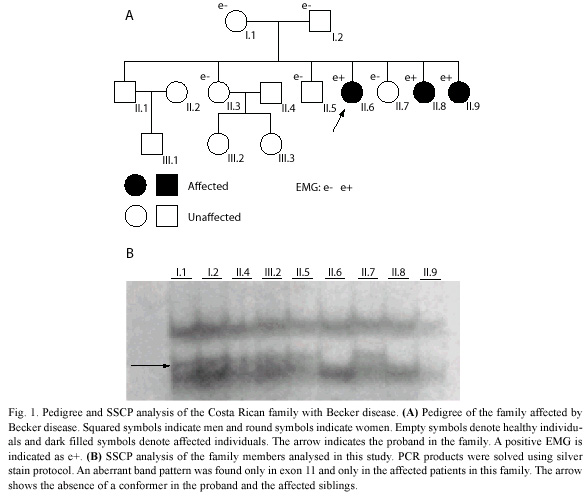
Patients: the study involved nine members of the family shown in Fig. 1A, in which the proband had been previously diagnosed with a myotonic condition, to know, myotonic dystrophy type 1 (DM1), but they were negative for the DM1 mutation (Morales et al. 2001). The proband (II.6, Fig. 1A), who was 20 years old at the time of this study, had failed to crawl by the time she was two years old. She experienced problems climbing stairs and her symptoms evolved into an important motor compromise. She developed limb distal muscle weakness, myotonia in tongue and hands, atrophy of the limbs, muscular contractures that made walking difficult, contractures in her hips and with a positive EMG, which detected typical myotonic discharges. The two affected siblings (II.8 and II.9, Fig. 1A) had similar symptoms to the proband, except milder and with an age of onset of about five years old. All of the family members were brought to the Costa Rican capital from their homes in order to accomplish more detailed clinical studies, which allowed us to establish a more accurate clinical diagnosis. Accordingly to our data, there is no known consanguinity in the family. Signed informed consent was obtained for all subjects for the clinical and molecular investigation in accordance with the ethical protocols approved by the Ethical Scientific Committee of the University of Costa Rica.
Clinical diagnosis: the clinical diagnosis was established after physical and electrophysiological tests. The EMG was carried out on eight members of the family and the slit lamp test was performed on two affected patients (II.8 and II.9), in order to exclude the possibility that they were affected by DM1. In addition to this, an EKG was done to all patients and the levels of CPK were also determined.
Clinical and electrophysiological examination: a complete neurological evaluation of all patients focused on muscles, analyzing the strength, the presence of the myotonic phenomenon before the muscular percussion and in the relaxation phase after a voluntary contraction. The muscular strength was recorded according the Medical Research Council scale. In addition, we developed a conventional and quantitative EMG study, with a motor neuroconduction study, including distal motor latency, motor nerve conduction velocities, F-M latencies and extent of the action potential of the median, ulnar, tibial and peroneal nerves. We also measured sensory nerve conduction velocities and sensory nerve action potentials of the right median, ulnar and sural nerves. All tests were performed at skin temperature of 31 ºC.
PCR of the CLCN1 exons: genomic DNA samples form nine members of the family, obtained from peripheral blood leukocytes were used for PCR amplification of the 23 exons covering the entire coding sequence of the CLCN1 gene. The intronic primers sets and PCR reaction conditions were described elsewhere (Lehmann-Horn et al. 1995). The experimental conditions were optimized for each primer. Each reaction had a final volume of 50 µl containing: 100 ng of DNA, 50 pmol of each PCR primer, 50 mM of each deoxynucleotide triphosphate, 10 mM Tris (pH 8.3), 50 mM KCL, 1.5 mM MgCl2 and 0.5 units of AmpliTaq polymerase. Amplification conditions were as follow: denaturation at 96 ºC for 5 min: 35 cycles at 96 ºC for 30 s, 55 ºC for 45 s, and 72 ºC for 1 min followed by a terminal elongation at 72 ºC for 5 min (Lehmann-Horn et al. 1995).
SSCP and sequencing: the PCR products of the 23 CLCN1 exons were analyzed for SSCP conformers as previously described (Orita et al. 1989). The PCR products were denatured and then loaded on 6 % nondenaturing polyacrylamide gels and electrophoresis was carried out as previously described (Nagamitsu et al. 2000). The products were detected using the silver stain protocol. Direct PCR sequencing was performed for the aberrant PCR product on the SSCP analysis. Samples were purified using Qiagen Spin-30 columns and both strands were sequenced by a dideoxi-termination method using the BDT (Big Dye Terminator) Sequencing Kit 3.0 (Perkin Elmer) and the ABI PRISM 377 DNA sequencer. Sequences were analyzed with the BioEdit 5.0.9 program (Ibis Therapeutics Carlsbad, CA92008).
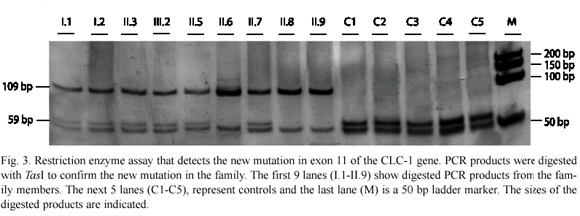
Allele-specific restriction digestion: TasI restriction sites were used for allele-specific restriction digestion of the exon 11 mutation in all family members and in 100 other samples from healthy individuals or with a disease other than MC, in order to confirm that the amino acid changed is the causing-disease mutation. The mutation abolishes the TasI restriction site generating size fragments of 50, 59 and 109 bp in heterozygous carriers and 50 and 59 pb bands in non-carriers of the mutation, thus the 109 bp fragment indicates the presence of the mutation. Genomic DNA (150 ng) from all of the samples was amplified for exon 11 using the conditions described above. Amplified products were digested with ten units of the restriction enzyme overnight according to the manufacturer instructions. The digestions were analyzed by electrophoresis in nondenaturing 5% acrylamide gel, using 0.6X Tris-borate/ EDTA (TBE) and bands were solved using the silver stain protocol. Gels were run at 200V for 3 h at room temperature.
Results
Clinical picture of the family members: the study was done when the family members were between 12 and 20 years old (mean of 15.6) with an age of onset of the main symptoms between 2 and 5 years of age (without taking into account the crawl problems in the first few months). The proband complained of difficulty in initial movements, on getting up in the mornings or after prolonged resting period, but after a while the movements improved (the warm up phenomenon). The three affected patients showed lessened reflexes, and the proband showed the steppage gait. Sensory examination was completely normal in all such family members and they showed clinical myotonia in different parts of their bodies. The three affected patients showed distal weakness, and two of them (II.6 and II.8) had also proximal weakness of the lower limbs with no pain at all in the affected patients. The proband also showed atrophy in the forearm and discreet peroneal atrophy. The rest of their relatives were normal. The slit lamp test performed in two affected members of the family was normal. Furthermore, they showed normal pupil reflexes. The EKG results were normal for all of the patients, and there was no family history of cardiac problems, arrythmias or other cardiac complication. CPK levels were mildly increased in the proband and in one of her sisters (II.9).
Electrophysiological examination: the EMG test was positive in the three affected patients, showing the classical myotonic runs and discharges together with the typical myophatic pattern. In the quantitative study, two of them (II.8 and II.9) showed a discrete diminution on the potentials amplitude with reduced duration, limited fibrillations, an early and complete recruitment with low amplitude in the full effort and with a normal polyphasia percentage. In the proband, the quantitative EMG showed motor unit potentials with high amplitude, duration and polyphasia percentage. Two patients (II.6 and II.8) presented diminished sensory conduction velocities and prolonged sensory distal latencies of the sural nerve. The rest of their relatives did not present electrical or clinical myotonia.
The molecular testing for myotonic dystrophy type 1 (DM1) was negative in this family (Morales et al. 2001). The phenotype was consistent with a clinical diagnosis of myotonia congenita, Becker disease. We carried out a further investigation of the CLCN1 gene by SSCP analysis.
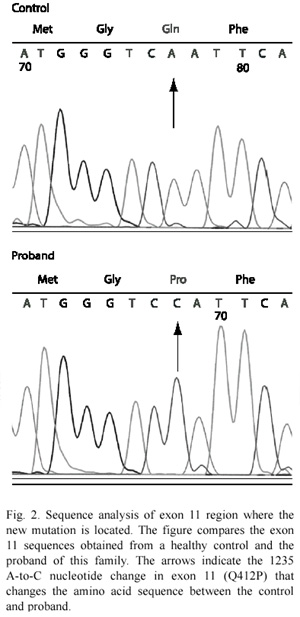
SSCP analysis, sequencing of aberrant SSCP conformers and allele-specific restriction digestion: after screening all of the 23 exons in the CLCN1 gene by SSCP, we found an aberrant SSCP conformer in exon 11 in the three affected patients (Fig. 1B) of the family in study (Fig. 1A). The band pattern observed in the SSCP analysis in the other members analysed of the family was the same as the control (Fig. 1B). Direct sequencing of the PCR product of exon 11 showed a new mutation, an A-to-C base change at nt 1235 (exon 11), which resulted in a substitution of glutamine for proline at codon position 412 (Q412P) (Fig. 2). Abolition of a TasI restriction site due to the A-to-C base change at nt 1 235 provided a quick assay for this new mutation in exon 11 (Fig. 3). TasI digestion generated fragments of 109 bp and 50 bp in the three affected patients who resulted homozygous for the new mutation (Fig. 3), and of 50 bp and 59 bp in normal healthy subjects (Fig. 3). Digestion of DNA with TasI in the other family members showed that all of them are heterozygous carriers of the new mutation. The assay in these samples generated the three expected bands for a heterozygous, at 109, 59 and 50 bp (Fig. 3). This new mutation was not found in 200 normal chromosomes. A comparison of CLC-1 channel sequences of various species showed that the glutamine at codon position 412 is highly conserved (Fig. 4).

Discussion
Inherited disorders that present myotonia as a major sign include DM1 and DM2, chloride channelopathies or myotonia congenita (Thomsen and Becker diseases) and sodium channelopathies (paramyotonia congenita, potassium-aggravated myotonia and hyperkalemic periodic paralysis) (reviewed in Morales and Cuenca 2004).
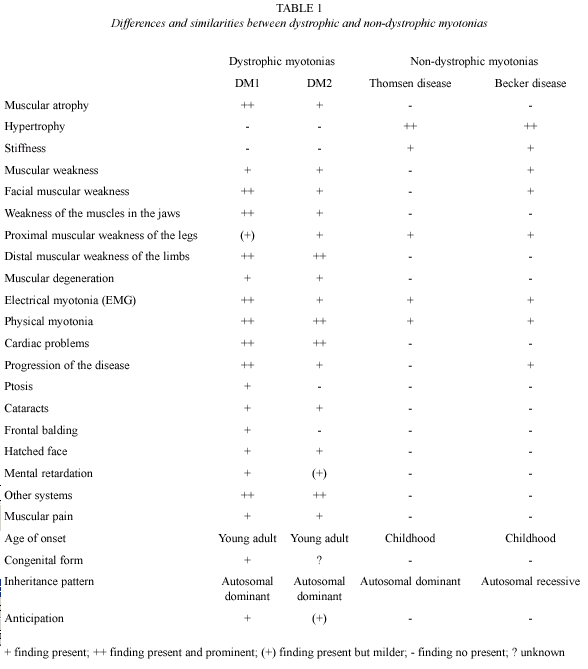
According to the clinical results obtained in this study, we concluded that the clinical picture of this family is compatible with myotonia congenita, and its autosomal recessive inheritance pattern suggested the diagnosis of Becker disease. This is the first clinical report in Costa Rica of a family affected with Becker disease, but the second regarding a non-dystrophic myotonic condition (Morales et al. 2003). The table 1 presents a comparison between myotonic dystrophy and myotonia congenita based on the information from Harper (2001), Koty et al. (1996), Zhang et al. (1996), Lehmann-Horn and Jurkat-Rott (1999), Meola (2000), and through the personal experience.
The molecular diagnosis obtained in this study confirmed the clinical diagnosis of this family, besides of the identification of a new mutation on the CLCN1 gene, enlarging the spectrum of mutations in this gene. The fact that the genotype of the affected patients correlates with their phenotypes, that the mutation was absent in 200 normal chromosomes and that the Q412P mutation affects a residue conserved among most members of the CLC channel family (Mailander et al. 1996) and among different species, suggest that this is the disease-causing mutation. Also, our data suggest that this is a rare mutation and probably restricted to the Costa Rican population. However, although our clinical data indicate the there is no consanguinity in this family, haplotype studies would be required in order to explore the possibility of identity-by-descents or of founder events for this mutation in the Costa Rican population. This would eventually suggest that this mutation is an ancestral mutation and that parents are, in some degree, related. Unfortunately, at the moment we do not have the haplotype analysis data, but it is something that needs to be done, not just for this mutation but also for the other ones we have been obtaining in other MC families (data not showed). The evidence that would confirm that this is the disease-causing mutation can be obtained through functional analysis studies of this new mutation, something that is expected to develop in a near future.
It is known that several mutations are associated with a particular inheritance pattern in the myotonia congenita. Some mutations are associated with the recessive form meanwhile others are associated with the dominant form. However, there are a few mutations that can behave as recessive or dominant, which is probably because of the genetic background in every patient (Meyer-Kleine et al. 1995, Koty et al. 1996, Pusch 2002, Colding-Jørgensen 2005). However, the relationships between different mutations and its inheritance pattern and a particular phenotype, both within and between a given set of different families having the identical mutation still requires clarification (Heatwole and Moxley 2007). The list of mutations already described in the CLCN1 gene is important as a starting point in the molecular characterization of the new families that appear in the future. The new mutation we report here, the Q412P behaves as recessive, which is consistent with the inheritance pattern and the phenotype in the family, where the members who were heterozygous for this mutation showed no myotonia, but the homozygous members were affected and showing the Becker phenotype. Nonetheless, for many mutations in the CLCN1 gene, it is still required to understand in a better way the genotype–phenotype relationships in MC in order to offer a proper clinical management and genetic counseling.
In 1996, Mailander et al. (1996), reported that clinically unaffected heterozygous males, but no females, presented latent myotonia in six families affected with Becker disease, however, our clinically unaffected heterozygous males (I.2 and II.4, Fig. 1A) did not show latent myotonia, meaning that not all of the mutations that cause the recessive form of the disease triggers latent myotonia, and this agrees with the fact that no other recessive mutation has been associated with latent myotonia. However, it is interesting to notice that our mutation (Q412P) is one amino acid away form another recessive mutation (F413C), first reported by Koch et al. (1992), then by Zhang et al. (2000a) who studied the functional consequences of this mutation and did not find the presence of latent myotonia, but also three amino acids away from one recessive mutation reported to cause latent myotonia (A415V) (Fig. 2) (Mailander et al. 1996). Thus, the ability of the mutation to cause latent myotonia could be intrinsic of the amino acid that is changed and that is capable to produce a stronger effect than the other mutations. For instance, in the A415V mutation, valine has considerably more bulk than alanine, something that does not happen with the other mutations, therefore producing a more steric effect that could affect the channel structure and in some way its function, even in the heterozygous state.
Members of the CLC family of voltage-gated chloride channels are found from bacteria to mammals with considerable degree of conservation in membrane-inserted, pore-forming region. The crystal structures of the CLC channels of Escherichia coli and Salmonella typhimurium provided a structural framework for the entire family. The CLC channels are homodimeric proteins with an overall rhombus-like shape. Each CLC dimer has two independent pores each contained within a single subunit, a so-called double barrel model. The CLC subunit consist of two roughly repeated halves that spans the membrane in opposite orientations. This antiparallele architecture defines a chloride selectivity filter within a 15-Å neck of an hourglass-shaped pore. Three Cl¯ binding sites within the selective filter stabilize ions by interactions with 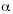 –helix dipoles and by chemical interactions with nitrogen atoms and hydroxyl groups of residues in the protein. The Cl¯ binding site nearest to the extracellular solution can be occupied either by a Cl¯ or by a glutamate carboxyl group where this glutamate work as a group that closes the pore by mimicking a Cl¯ ion (Dutzler 2004a). The channels have a complex gating behaviour, in which channel opening and closing are tightly coupled to ion permeation. In the closed conformation of the channel, the conserved glutamate residue occupies one of the ion-binding sites; on the channel opening, this glutamate residue moves out of the binding site, thereby making room for an additional Cl¯ ion ( Dutzler et al. 2003, Dutzler 2004b).
–helix dipoles and by chemical interactions with nitrogen atoms and hydroxyl groups of residues in the protein. The Cl¯ binding site nearest to the extracellular solution can be occupied either by a Cl¯ or by a glutamate carboxyl group where this glutamate work as a group that closes the pore by mimicking a Cl¯ ion (Dutzler 2004a). The channels have a complex gating behaviour, in which channel opening and closing are tightly coupled to ion permeation. In the closed conformation of the channel, the conserved glutamate residue occupies one of the ion-binding sites; on the channel opening, this glutamate residue moves out of the binding site, thereby making room for an additional Cl¯ ion ( Dutzler et al. 2003, Dutzler 2004b).
Our mutation and others in the same region are located between the transmembrane –helix domains K and L (Dutzler et al. 2002), region that has not been properly characterized; only the filter selective has been largely characterized (Dutzler 2004a, Dutzler 2004b). The only mutation functionally characterized on this region is the F413C (Zhang et al. 2000a). Zhang et al. (2000a) developed such a study and they found that this mutation could lead to a decrease in the macroscopic chloride currents and hyperexitability in the skeletal muscle membranes. The significance of this change for myotonia congenital, however, is uncertain. They also propose that this mutation may affect folding, assembly or trafficking of CLCN1 channel proteins. It is important to expand these studies to examine other mutations in this domain. By comparing the phenotypes caused by each mutation it may be possible to determine the specific function of this domain in the protein.
Perhaps some domains are more important because they stabilize the channel to allow a proper Cl¯ conductance. Mutations at or near the selective filter for example, could have a major impact on the Cl¯ conductance. Grunnet et al (Grunnet et al. 2003) reported two dominant mutations that show this effect. They propose that the M128V mutation appears to be located in the homomeric interaction domain of CLCN1, and therefore is suspected to influence the dimerization. Further, the E193K mutation, which is located next to the selective filter, introduces a selective shift in the CLCN1 channel, which may explain the altered selectivity of this mutant. Both mutations behave in a dominant manner with a rightward shift in the currentvoltage relationship, which results in reduced CLCN1 currents at physiological relevant potentials. Thus, it is possible that the inheritance pattern of some mutations in the CLCN1 gene can be explained by the position of the mutation and their effect in the function-structure relationship of the most important domains of the channel that regulate the Cl¯ conductance.
The family we report here seems to represent a typical example of Becker disease: non-progressive, non-disabling and probably not as severe as other families affected with this disease. However, by studying the functional consequences of this new mutation, we may be able to provide a better understanding of the phenotype of the affected members. This could provide an explanation for the diminution of the sensitive conduction velocities and prolonged sensory distal latencies and other features in this family. It may also be possible to find a function for the region where the mutation is and to establish a better phenotypegenotype correlation, which could help us to better understand the structure-function-genotype relationships of the different mutations in the CLCN1 gene.
Acknowledgments
The authors thank the family members for their participation in this study. We also thank Fernando Ortiz and Zaida Gutierrez for the technical support and Jim Hilley for his comments, corrections and reviewing on this paper. The IAEA, DAAD and the University of Costa Rica supported this work.
Resumen
La miotonía congénita es una enfermedad muscular caracterizada por miotonía, hipertrofia y rigidez. Se presenta con dos patrones de herencia, autosómica dominante en cuyo caso recibe el nombre de miotonía de Thomsen, o autosómica recesiva conocida como miotonía de Becker. En este trabajo se confirmó el diagnóstico clínico presuntivo hecho hace algunos años en una familia con una condición miotónica y se reporta una nueva mutación en el gen CLCN1. El diagnóstico clínico se estableció después de estudios oculares, cardíacos, neurológicos y electrofisiológicos. El diagnóstico molecular fue hecho mediante la PCR, SSCP y secuenciación del gen CLCN1. El caso índice y los otros individuos afectados exhibieron debilidad muscular proximal y distal, pero no se encontró hipertrofia ni dolor muscular. Los reflejos miotáticos estuvieron disminuidos y la sensibilidad fue normal. Se encontró miotonía clínica y eléctrica solo en los individuos afectados. Las pruebas de lámpara de hendidura y electrocardiograma resultaron normales. Dos individuos afectados presentaron disminución de las velocidades de conducción sensitiva y latencias distales sensoriales prolongadas. El cuadro clínico concuerda con la miotonía de Becker, lo cual se confirmó con el hallazgo de una mutación responsable de la enfermedad en el gen CLCN1 (Q412P), la cual se encontró en la familia y estuvo ausente en 200 cromosomas provenientes de la población general. No se encontró miotonía latente, por lo que probablemente la habilidad de causar este signo subclínico es intrínsica de cada mutación. Afinar el diagnóstico clínico diferencial de las enfermedades neuromusculares permitiría enfocar los estudios moleculares hacia la confirmación del diagnóstico inicial en forma eficiente, lo cual permitiría un manejo clínico y asesoramiento genético más adecuados y una mejora en la calidad de vida de los pacientes y sus familias.
Palabras clave: miotonía congenita, distrofia miotónica, miotonía de Becker, canalopatía de cloruro, SSCP.
References
Colding-Jorgensen, E. 2005. Phenotypic variability in myotonia congenita. Muscle Nerve 32: 19-34. [ Links ]
Dutzler, R. 2004a. Structural basis for ion conduction and gating in ClC chloride channels. FEBS Lett. 564: 229-233. [ Links ]
Dutzler, R. 2004b. The structural basis of ClC chloride channel function. Trends Neurosci. 27: 315-320. [ Links ]
Dutzler, R., E.B. Campbell, M. Cadene, BT. Chait & R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 415: 287-294. [ Links ]
Dutzler, R., E.B. Campbell & R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science 300: 108-112. [ Links ]
Esteban, J., A.M. Neumeyer, D. McKenna-Yasek & R.H. Brown. 1998. Identification of two mutations and a polymorphism in the chloride channel CLCN-1 in patients with Beckers generalized myotonia. Neurogenetics 1: 185-188. [ Links ]
Grunnet, M., T. Jespersen, E. Colding-Jorgensen, M. Schwartz, D.A. Klaerke, J. Vissing, S.P. Olesen & M. Duno. 2003. Characterization of two new dominant ClC-1 channel mutations associated with myotonia. Muscle Nerve 28: 722-732. [ Links ]
Harper, P.S. 2001. Myotonic Dystrophy. W.B. Saunders, London, England. [ Links ]
Heatwole, C. & R.T. Moxley III. 2007. The nondystrophic myotonias. Neurotherapeutics 4: 238-251. [ Links ]
Jurkat-Rott, K., H. Lerche & F. Lehmann-Horn. 2002. Skeletal muscle channelopathies. J. Neurol. 249: 1493-1502. [ Links ]
Koch, M.C., K. Ricker, M. Otto, F. Wolf, B. Zoll, C. Lorenz, K. Steinmeyer & T.J. Jentsch. 1993. Evidence for genetic homogeneity in autosomal recessive generalised myotonia (Becker). J. Med. Genet. 30: 914-917. [ Links ]
Koch, M.C., K. Steinmeyer, C. Lorenz, K. Ricker, F. Wolf, M. Otto, B. Zoll, F. Lehmann-Horn , K.H. Grzeschnik & T.J. Jentsch. 1992. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 257: 797-800. [ Links ]
Koty, P.P., E. Pegoraro, G. Hobson, H.G. Marks, A. Turel, D. Flagler, M. Cadaldini, C. Angelini & E.P. Hoffman. 1996. Myotonia and the muscle chloride channel: dominant mutations show variable penetrance and founder effect. Neurology 47: 963-968. [ Links ]
Lehmann-Horn, F. & K. Jurkat-Rott. 1999. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 79: 1317-1372. [ Links ]
Lehmann-Horn, F., V. Mailander, R. Heine & A.L. George. 1995. Myotonia levior is a chloride channel disorder. Hum. Mol. Genet. 4: 1397-1402. [ Links ]
Mailander, V., R. Heine, F. Deymeer & F. Lehmann-Horn. 1996. Novel muscle chloride channel mutations and their effects on heterozygous carriers. Am. J. Hum. Genet. 58: 317-324. [ Links ]
Meola, G. 2000. Clinical and genetic heterogeneity in myotonic dystrophies. Muscle Nerve 23: 1789-1799. [ Links ]
Meyer-Kleine, C., K. Steinmeyer, K. Ricker, T.J. Jentsch & M.C. Koch. 1995. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am. J. Hum. Genet. 57: 1325-1334. [ Links ]
Morales, F., P. Cuenca, R. Brian, M. Sittenfeld & G. del Valle. 2001. Diagnóstico molecular de la Distrofia Miotónica (DM) en Costa Rica. Acta Méd. Cost. 43: 159-167. [ Links ]
Morales, F., P. Cuenca, G. Del Valle G, R. Brian, M. Sittenfeld, O. Montoya, T. Ashizawa, A. Rosa & K. Johnson. 2003. Miotonía congénita: caracterización clínica de una familia costarricense afectada por la Enfermedad de Thomsen. Neuroeje 17: 82-86. [ Links ]
Morales, F. & P. Cuenca. 2004. Aspectos genéticos y moleculares de las enfermedades miotónicas. Rev. Neurol. 38: 668-6674. [ Links ]
Nagamitsu, S., T. Matsuura, M. Khajavi, R. Armstrong, C. Gooch , Y. Harati & T. Ashizawa. 2000. A "dystrophic" variant of autosomal recessive myotonia congenita caused by novel mutations in the CLCN1 gene. Neurology 55: 1697-1703. [ Links ]
Orita, M., Y. Suzuki, T. Sekiya & K. Hayashi. 1989. Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5: 874-879. [ Links ]
Pusch, M. 2002. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 19: 423-434. [ Links ]
Sun, C., L. Tranebjaerg, T. Torbergsen, G. Holmgren & M. Van Ghelue. 2001. Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur. J. Hum. Genet. 9: 903-909. [ Links ]
Zhang, J., S. Bendahhou, M.C. Sanguinetti & L.J. Ptacek. 2000a. Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology 54: 937-942. [ Links ]
Zhang, J., A.L. George, Jr., R.C. Griggs, G.T. Fouad, J. Roberts, H. Kwiecinski, A.M. Connolly & L.J. Ptacek. 1996. Mutations in the human skeletal muscle chloride channel gene (CLCN1) associated with dominant and recessive myotonia congenita. Neurology 47: 993-998. [ Links ]
Zhang, J., M.C. Sanguinetti, H. Kwiecinski & L.J. Ptacek. 2000b. Mechanism of inverted activation of ClC-1 channels caused by a novel myotonia congenita mutation. J. Biol. Chem. 275: 2999-3005. [ Links ]

