










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.52 n.3 San José Sep. 2004
Expression of the rice hoja blanca virus (RHBV) non-structural protein 3 (NS3) in Escherichia coli and its in situ localization in RHBV-infected rice tissues
Miguel Muñoz1 , Isela Bolaños1 , Griselda Arrieta-Espinoza1 & Ana M. Espinoza1,2
1 Centro de Investigación en Biología Celular y Molecular (CIBCM), Universidad de Costa Rica.
2 Escuela de Agronomía, Facultad de Ciencias Agroalimentarias, Universidad de Costa Rica.
Received 20-VIII-2002. Corrected 09-VI-2003. Accepted 30-VII-2003.
Abstract
The non-structural NS3 protein gene from the rice hoja blanca virus (RHBV) was fused to the glutathione- S-transferase carboxilic end and expressed in Escherichia coli strain JM83. Large quantities of fusion protein were produced in insoluble form. The fusion protein was fractionated in SDS-PAGE and purified by electroelution, polyclonal antibodies were raised in rabbit and the antiserum was absorbed with bacterial crude extract. A band of similar size as that of NS3 protein was observed in Western blots using extracts from RHBV-infected rice plants. Immunoelectron microscopy with colloidal gold-labeled antibodies against NS3 protein and the viral nucleocapsid protein revealed in situ accumulation of NS3 protein in the cytoplasm but not in the viral inclusion bodies, vacuoles or chloroplasts of RHBV-infected plants, following the same pattern of distribution as the RHBV nucleocapsid protein. Rev. Biol. Trop. 52(3): 765-775. Epub 2004 Dic 15.
Key words: Rice hoja blanca virus, RHBV, NS3, polyclonal antibodies, immunoelectron microscopy.
Palabras clave: Virus hoja blanca, arroz, RHBV, NS3, anticuerpos policlonales, inmunoelectro microscopía.
Rice hoja blanca is a viral disease that occurs in cyclical outbreaks, reaching epidemic levels maintained over several years. It is currently affecting rice production in Tropical America and the Caribbean. Hoja blanca disease is caused by a planthopper-transmitted virus: the rice hoja blanca virus (RHBV, Tenuivirus), which induces chlorotic stripes, systemic chlorosis and seed sterility in cultivated rice plants (Jennings 1963). The virus is persistent and propagative in the delphacid insect vector (Tagosodes orizicolus, Homoptera: Delphacidae), which in turn is able to transmit the virus transovarially to the progeny (Nault and Ammar 1989).
RHBV is a non-enveloped multipartite RNA virus, it has an 18 Kb-genome arranged in four negative sense and ambisense genomic RNAs of different sizes, totaling seven open reading frames (ORFs) (Ramírez and Haenni 1994). Sequence analysis of RHBV RNAs has revealed a viral RNA polymerase ORF in RNA-1 similar to rice stripe virus RNA polymerase (Toriyama et al. 1994, Muñoz et al., unpublished results). Two ORFs encoding putative membrane-glycoproteins of unknown function are found in the ambisense RNA-2 (de Miranda et al. 1995, 1996). The ambisense RNA-3 encodes the viral nucleocapsid protein (N) and a non-structural protein (NS3) of unknown function (de Miranda et al. 1994). The ORFs for the inclusion bodies protein and another non-structural protein (NS4), of unknown function are located in the ambisense RNA-4 (Ramírez et al. 1993).
Genomic RNA-3 and RNA-4 are the most abundant RNAs in RHBV-infected tissues, however, only two genes have been asigned to known proteins: N-protein, found associated with ribonucleoproteins (RNP), and the non-capsid protein (NCP), found in cytoplasmic inclusion bodies. Sequence analysis has revealed short stretches of aminoacids in NS4 similar to those found in potyviral insect transmission factors (Ramírez and Haenni 1994). The function of the ns3 gene is still unknown, DNA database searches using ns3 sequence from different tenuiviruses has not revealed homology sequence so far (Huiet et al. 1991, Kakutani et al. 1991, de Miranda et al. 1994). It has been suggested that it may encode the viral movement protein (Ramírez and Haenni 1994).
Infectious clones of RHBV or mutant versions of the ns3 gene are not yet available for functional analysis, however, it may be possible to study the subcellular localization of NS3 protein using specific antibodies and derive a model for further study. This strategy requires the purification of the viral proteins and the production of polyclonal antibodies. The viral gene can be subcloned in bacterial expression vector systems, by insertion at the 3 end of a gene that encodes a highly expressed transporter protein (Smith and Corcoran 1992), producing a chimeric fusion protein (Marston 1987). One of such expression vectors is the pGEX plasmid family, the polypeptide of interest is fused to the carboxilic end of the 26 kDa glutathione-S-transferase (GST) from Schistosoma mansoni (Smith et al. 1986). These vectors contain the IPTG-inducible tac promoter (Amann et al. 1983), and the GST open reading frame (ORF) (Smith et al. 1986), in which the termination codon is replaced by a multiple cloning site followed by the termination codon TGA. Protein expression is induced with isopropyl thio-b-D-galactoside (IPTG), allowing the purification of the fusion protein from crude bacterial lysates under non-denaturing conditions by glutathione affinity chromatography (Smith and Cocoran 1992).
To determine in situ localization of the NS3 protein in RHBV-infected plants, the NS3-GST fusion protein was expressed in E. coli, purified and used for the production of polyclonal antibodies. Immunological tests revealed the expression of the NS3 protein in extracts of infected plants; gold-labeled antibodies localized the protein in the cytoplasm along with the viral nucleocapsid.
Materials and methods
Cloning and expression of the RHBV ns3 gene in E. coli: The ns3 gene was amplified by PCR from a RHBV cDNA-3 full-length clone (Genebank L07940) previously sequenced from a viral genomic library (de Miranda et al. 1994). A 0.61 Kb fragment containing the full ORF was recovered using two specific primers. NS3-Forward primer (5´-ATCGATGGATCCAAATGAACGTGTC-CTTTTGAA- 3) includes the 5´ end of the ORF, (bases 96 to 115, in italics), the BamH1 restriction site (bold) and the initiation codon (underlined). The NS3-Reverse primer (5-AGAGAGGAATTCACTA GAAT G -GAAGCAGTAA-3) contains the 3´ of the ORF (bases 717 to 734, in italics), the EcoR1 restriction site (bold) and the termination codon (bold italics) (Fig. 1). Ten nanograms (ng) of plasmid template were amplified by PCR in a mixture containing 20 mM Tris-HCl pH 7.8, 40 mM KCl, 1.5 mM MgCl2 , 0.1% BSA, 0.2 mM dNTPs, 0.4 µM each primer and 2.5 units of Taq polymerase. The PCR thermal profile was two cycles of 94ºC for 60 s, 45ºC for 30 s and 72ºC for 2 min, followed by 32 cycles of 94ºC for 60 s, 55ºC for 30 s and 72ºC for 2 min. The PCR product was digested with BamHI and EcoRI enzymes and inserted inframe at the 3end of the GST gene in the pGEX3X expression vector, to make plasmid pGST-NS3. The insertion site is located down-stream the factor Xa protease recognition site (Ile-GLu-Arg) for cleavage of the protein from the fusion product. Heat-shock competent E. coli cells, strain JM83, were transformed with pGST-NS3 and pGX3X vector. Bacterial cultures were grown for two hours in 50 ml liquid LB broth, then IPTG (0.1 mM) was added and the culture was incubated for six hours at 37°C. Induced cells were separated by centrifugation 10 000 rpm for 5 min and re-suspended in 10 ml saline phosphate buffer 1X PBS. Cells were lyzed using three different methods: thermal shock by freezing at -70°C for 15 min and thawing at 37°C for 20 min (four cycles), enzymatic digestion with lysozyme (120 mM) or four pulses of sonication for 30 s (Virsonic 50, Virtis, USA). The lysate was centrifuged 10 000 rpm for 5 min at 4°C, the supernatant was transferred to a separate tube and the pellet was re-suspended in 1X PBS, 1% Triton X-100 (SIGMAÒ ), 50 mM EDTA, 1 mM PMSF (phenylmethylsulfonylfluoride), and 10 mM DTT (dithiothreitol). The supernatant and pellet were fractionated in 12% SDS-PAGE (Laemmli 1970). Since the fusion protein was mainly insoluble, glutathione affinity chromatography columns were clogged and little protein was recovered. When solubilized protein was fractionated, co-purification of large quantities of other bacterial protein was recovered. Therefore, crude extracts were fractionated in 12% SDS-PAGE preparative gel and stained in 0.1% Coomassie blue. The fusion protein band was cut out and electroeluted for two hours at 200 mA in Tris-Glycine buffer (25 mM Tris-base, 250 mM glycine and 0.1% SDS), using an electroelution chamber (BioRad " ). The purified fusion protein was dialyzed with 1 X SSC and analyzed in SDS-PAGE gels to estimate protein concentration.
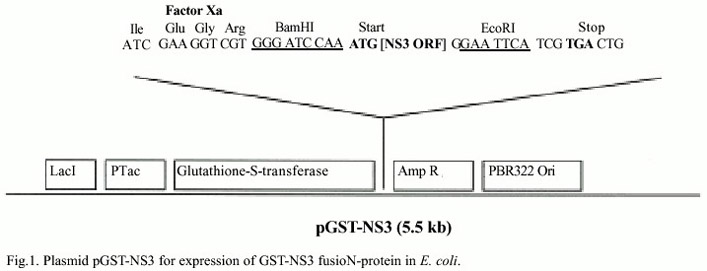
Preparation of polyclonal antibodies against the GST-NS3 fusion protein: Polyclonal antibodies directed against the GST-NS3 fusion protein were produced in rabbits by injection into the hind foot with an emulsion of 0.5 ml Freunds complete adjuvant and 0.5 ml of purified protein (approximately 1.66 mg/ml) or bacterial crude extract containing the GST-NS3 fusion protein. Two weeks later a similar amount of antigen plus Freunds incomplete adjuvant was injected, another four booster inoculations were injected, once every 2 weeks. Rabbits were bled from the ear 12 days after the last inoculation, blood was collected and incubated two hours at 37°C, then incubated at 4°C overnight and the antiserum was separated by centrifugation at 3000 g for 30 min. The antiserum was evaluated by double immunodiffusion test in agar and in Western blots. Immunodiffusion was done in 0.3% agar plates, diluting two-fold the antigen and antiserum, followed by incubation for 24 to 48 hours in a moist chamber.
Western blot analysis: Western blot assays were carried out as indicated in Sambrook et al. (1989). Crude extract of E. coli and rice plants, as well as RHBV-infected plants and RHBV proteins (22 kDa NCP and 34 kDa N-protein) were used as controls. Leaf tissue (0.2 g) was homogenized in liquid nitrogen and mixed with 1 ml of sample buffer for protein electrophoresis (0,065 M Tris-HCl pH 6.8, 2% SDS, 40% glycerol, 5% mercaptoethanol and 0,001% bromophenol blue) supplemented with 50 mM EDTA, 10 mM DTT and 1 mM PMSF. The membrane was incubated first with the antiserum against crude or the purified extract (1/1000 dilution), and then with alkaline phosphatase-labeled antirabbit IgG (1/500 dilution) (SIGMA). Nitroblue tetrazolium (NBT) and 5-bromo-4- chloro-3-indolyl phosphate (BCIP) were used as substrates. Antiserum against purified extract was further purified by absorption with 10 mg of freeze-dried crude extract from E. coli expressing the pGX3X without the ns3 gene and incubated for two hours at 37°C. The IgG fraction was purified through affinity chromatography. Staphylococcus Protein A-Sepharaised CL-4B column (Pharmacia®) was washed with glycine buffer (0.2 M glycine-HCl and 0.5 M NaCl, pH 2.8) then it was equilibrated with Trizma Base buffer (0.05 M Trizma Base and 0.5 M NaCl, pH 7.5). Samples of 3 ml were added to the column and filtered for 45 minutes. The column was washed with Trizma Base buffer and the flow-through was discarded, the IgG fraction was eluted with Glycine buffer and samples were collected and neutralized to pH 7 with Trizma Base pH 8.8. The column was washed with Trizma Base buffer pH 7.5 and the samples were dialyzed with PBS 1X. Protein concentration was estimated by spectrophotometry at 280 nm.
Immunogold labeling of ultrathin sections of infected leaves. Leaf tissues from healthy and infected plants were fixed in 2.5% glutaraldehyde in 0.05 M sodium cacodylate-HCl, pH 7.2 for 16 hr at room temperature. The samples were dehydrated in a series of ethanol solutions (30-100%) for 20 min at room temperature. Propylene oxide was gradually substituted for ethanol (3:1, 2:2, 1:3, and two changes of 100% propylene oxide). Infiltration was done with Spurr resin in several steps (3:1, 2:2, 1:3, and two changes of 100% resin) one hour per step. Polymerization carried out at 60°C for 24 hr. Samples were sectioned in an ultramicrotome LKB 8800 and the resin was removed by incubation in a saturated solution of sodium metaperiodate for 10 min in the dark and then washing with distilled water three times for five minutes in the dark. Nickel grids (200 mesh) were placed onto a droplet of a 1:50 dilution of rabbit anti-NS3 antibody and 1:250 dilution of mouse anti-N-protein antibody in blocking buffer (200 mM Tris-HCl pH 7.4, 1% BSA, 0.1% gelatin and 1% Tween 20) for 8 hr at 4°C and then washed for 1 min in blocking solution. Grids were placed onto a droplet containing gold-labeled antibodies (AuroProbe EM GAR G10, Janssen, and diluted 1:20 in blocking buffer) for 2 hr at room temperature. Adouble labeling was done using a mixture of antimouse IgG and protein A-colloidal gold of three diameters (10 and 20 nm antirabbit and 5 nm antimouse). Two antisera were used: mouse anti-N-protein, diluted 1:250, and rabbit anti-NS3, diluted 1:50. The sections were then stained with 4% uranyl acetate for 15 min, rinsed with distilled water and then stained with lead citrate for 15 min. The preparations were examined using a Hitachi 7000 electron microscope.
Results
Expression induction and analysis of the GST-NS3 fusion protein: Accumulation of a 49 kDa protein was observed upon induction with IPTG in E. coli cultures containing plasmid pGST-NS3. A 49 kDa protein is expected for a fusion derived from the 26 kDa GST and 23.2 kDa NS3 protein. Non-induced control sample did not accumulate the GST-NS3 fusion protein at all. The GST-NS3 fusion protein was found not to be toxic in E. coli, since post-induction periods as long as 6.5 hrs at 37ºC were tested without significant decrease in protein accumulation. Little degradation was observed in SDS-PAGE, indicating that the fusion protein is stable when expressed in E. coli (data not shown), however , protein additives were added to the lysis buffer: a serine protease-inhibitor (PMSF), a chelating agent (EDTA) and antioxidant (DTT) to increase protein stability. Furthermore, a non-ionic surfactant (1% Triton X-100) was added to diminish the association of the fusion protein to other bacterial proteins (Smith and Corcoran 1992). Cells were lysed enzymatically (lysozyme) or mechanically (sonication and multiple freeze-thawing cycles). Disruption of bacterial cells by sonication was the most efficient and reproducible method of protein extraction, total cell lysis was achieved with no protein degradation (data not shown).
Upon cell disruption, both the supernatant and the insoluble fraction were analyzed in 12% SDS-PAGE. The GST-NS3 fusion protein was always found in the insoluble pellet along with bacterial proteins (data not shown), addition of solubilizing agents such as Triton X-100 increased its solubility; however other bacterial proteins were also observed in the supernatant along with the GST-NS3 fusion protein. Although pGEX vectors allow the protein of interest to be purified under non-denaturizing conditions through affinity chromatography (Smith and Corcoran 1992), the eluted fusion protein was highly contaminated with other bacterial proteins that were bound to the column in a non-specific way. Thus, the fusion protein was purified under denaturing conditions using SDS-PAGE and electroelution, as suggested by Marston (1986).
Serological analysis of the GST-NS3 fusion protein: Rabbit antiserum produced against crude extract from transformed E. coli and against the purified fusion protein were able to detect denatured GST-NS3 proteins in Western blots, even at antisera dilutions of 1/1000 (Fig. 2 a and b). Although we expected the antisera raised against the purified extract to have higher specificity than that of crude extract, the GST-NS3 polyclonal antibodies also reacted non-especifically against a large collection of E. coli proteins (Fig. 2a, lanes 1 through 3). However, both GST and GST-NS3 were clearly recognized by the antisera (lane 2 and 3, respectively). Since Mycobacterium was included in the adjuvant mixture for immunization, the antibodies raised may have cross-reacted with conserved proteins or epitopes in both E. coli and Mycobacterium. Plant proteins were not detected (lane 6), while RHBV-infected plants showed a single band of 23 kDa (lane 5), as expected for the NS3 protein. Other viral protein, such as the NCP (lane 7), were not recognized by the antisera, however, a non-especific reaction was observed with the purified viral ribonucleoproteins (lane 8), although the band was slightly larger than expected for the N-protein (34 kDa).
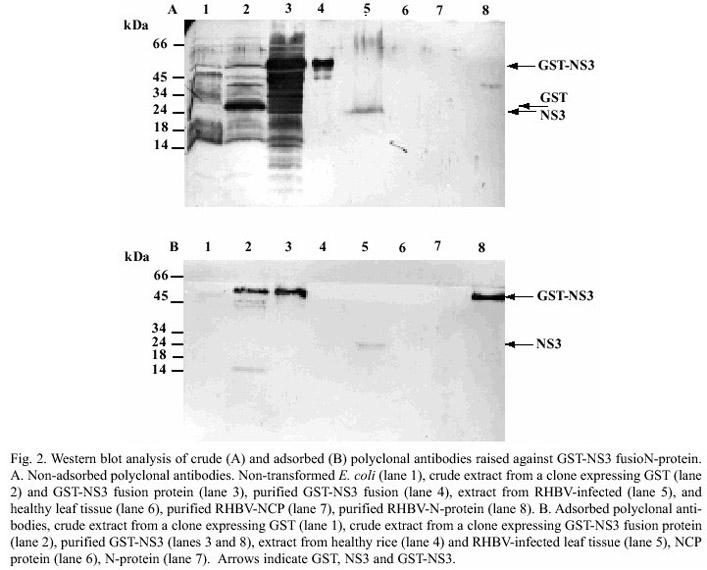
Antiserum specificity was increased by adsorption of antibodies with lyophilized crude extracts from E. coli expressing GST (Fig. 2b). This procedure reduced non-specific binding with proteins other than GST-NS3 in bacteria (compare lanes 1 and 2), whereas both the GST-NS3 and RHBV-NS3 were detected in crude extracts from induced-E. coli cultures containing pGST-NS3 (lane 2), purified fusion protein (lanes 3 and 8) and extract from RHBV-infected leaves (lane 5). Furthermore, no signal was observed in extracts from healthy rice plants (lane 6), neither from viral proteins N and NCP (lanes 6 and 7 respectively). The molecular weight of GST and NS3 proteins are similar (26 and 23 Kda, respectively), however, no signal was observed when extracts of E. coli expressing GST alone were analyzed by Western blot (Fig 2b. lane 1). This indicates that the adsorption procedure was efficient in precipitating non-specific antibodies, including those against GST, and that antibodies against NS3 do not cross react with epitopes from GST.
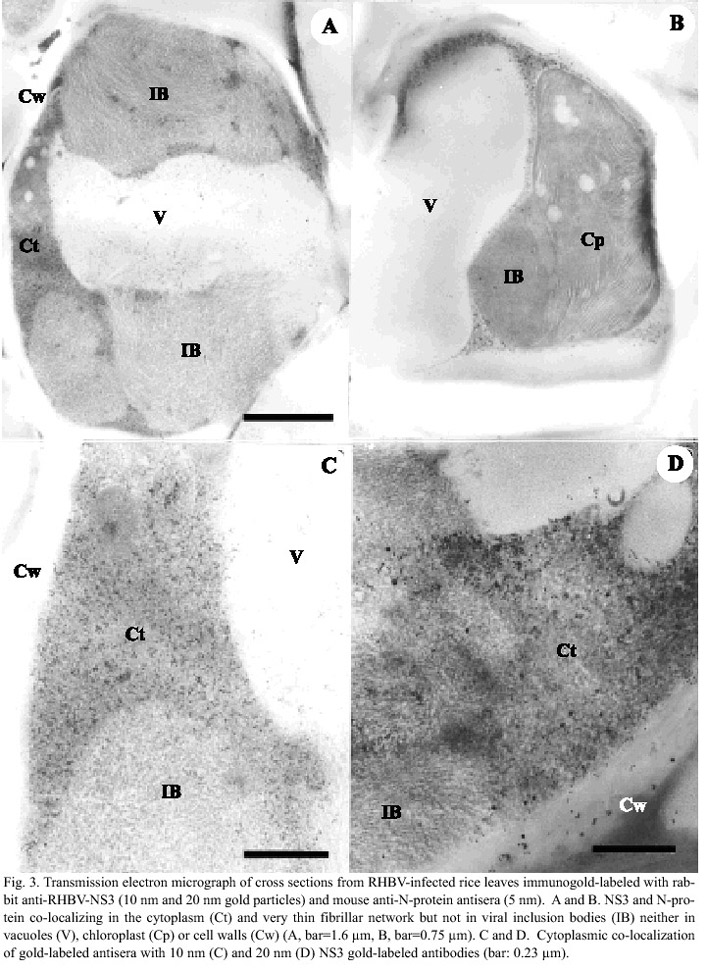
Immuno-localization of NS3 protein in RHBV-infected rice tissues: Polyclonal NS3 antibodies were purified and the IgG fraction was labeled with colloidal gold (20 nm and 10 nm gold particles); healthy and RHBV-infected rice leaves were analyzed in combination with mouse antiviral ribonucleoprotein (N-protein)antibodies labeled with 5 nm colloidal gold. While no signal was observed in mesophyll cells of healthy plants (data not shown), the antibodies mixture localized both NS3 and N-protein in infected cells (Fig. 3a and 3b). Both proteins were found in the same subcellular locations: mainly in the cytoplasm (Ct) and collapsing vacuoles (Fig. 3a). The antibodies could not detect either N-protein or NS3 in chloroplast (Cp), cell walls (Cw), vacuoles (V) or inclusion bodies (IB) (Figs. 3a and 3b). Gold-labeled N-protein-antibodies were more abundant than NS3 labeled antibodies (Fig. 3c), a pattern that is more clear when larger gold particles were used (20 nm, Fig. 2d). The pattern of accumulation of labeled antibodies against NS3 and N-protein resembled that of the viral nucleocapsid protein in mesophyll cells, as previously described by Espinoza et al. (1993).
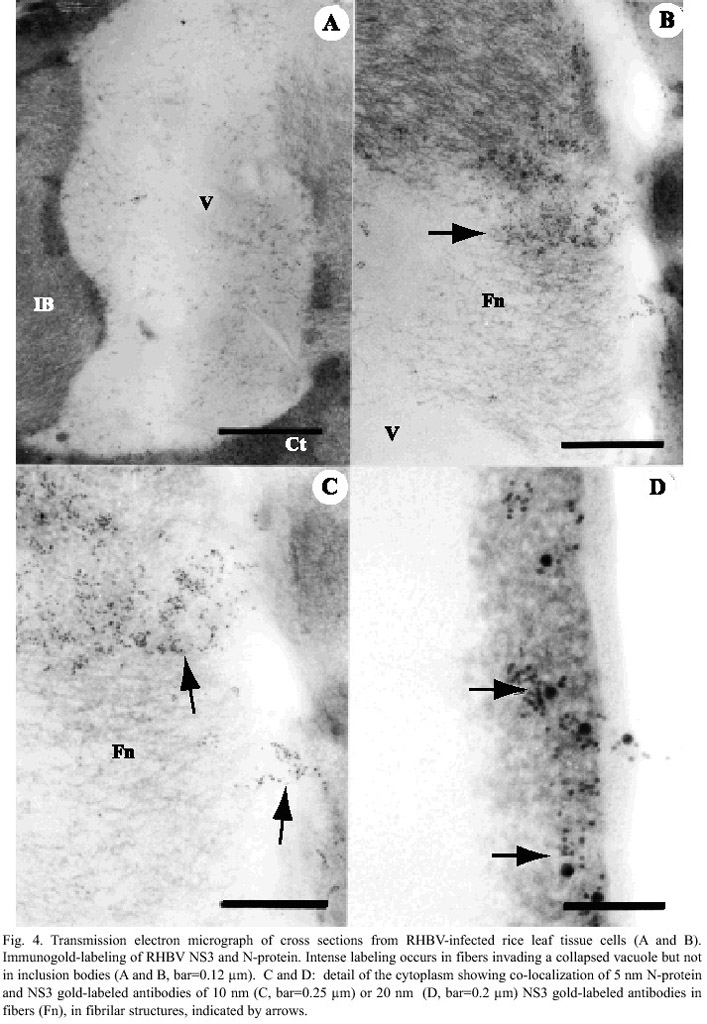
Cells from infected tissue showed collapsed vacuoles and a network of loosely attached fibrilar structures that extend towards the space left by the vacuole (Fig. 4a). Interestingly, antibodies raised against both NS3 and N-protein were also found associated to thin fibers (Fig. 4b and 4c), especially in collapsing cells that has already accumulated large masses of viral inclusion bodies. Although the fibrilar network was no evenly coated with labeled antibodies, most of the fibers were coated with both NS3 and N-protein antibodies (Fig. 4c). N-protein antibodies were observed at higher frequency as compared to those of NS3 (Fig. 4c and 4d). These structures were also observed in the cytoplasm.
Discusion
The function of viral proteins can be studied using different approaches including mutagenesis and complementation with wild type genes, ectopic expression of wild type proteins in transgenic plants or chimeric proteins containing reporter peptides, such as fluorescent domains. However, because of the lack of infectious clones and the requirement of planthopper vectors for virus transmission, mutagenesis experiments have not been carried out with tenuiviruses. Moreover, the requirement of full-cDNAs, in vitro transcription of each one of the genomic RNAs and mix-and-match of each component along with the viral replicase makes this sort of experiments technically very difficult to perform.
An alternative approach is the expression and purification of viral genes as fusion proteins expressed in bacteria and the production of antiserum for immunological studies. However, when recombinant proteins are over-expressed in E. coli, they often accumulate in the form of insoluble inclusion bodies, especially when the size of the fusion protein is over 50 kDa (Marston 1987), which is close to the size of the GST-NS3 protein. Insolubility is associated with the presence of extensive and highly hydrophobic regions; as a result polypeptides tend to aggregate in inclusion bodies in order to minimize exposure of hydrophobic amino acid residues in aqueous solution (Volkin and Klibanov 1989). Modifications, such as thio-disulphide interchange and molecular conformational changes, also lead to exposure of hydrophobic amino acid residues and protein aggregation. Although insoluble proteins aggregate can be partially solubilized with non-ionic detergents, other bacterial proteins may contaminate the extract, inducing the production of other antibodies besides the ones against the protein of interest, which reduces specificity of the antiserum.
Antibodies against the RHBV ribonucleo-protein and the NS3 protein colocalized exclusively in the cytoplasm, no labeling was observed in vacuoles, chloroplasts, cell walls or viral inclusion bodies. The fact that NS3 and N-protein were not detected in the inclusion bodies, agrees with the observation by Espinoza et al. (1993), that inclusion bodies are composed by a single kind of protein (non-capsid protein or NCP). It also rules out the function of the NS3 as a component of the inclusion bodies.
It is intriguing that both NS3 and the N-protein co-localized in the same subcellular compartment. In nearly collapsed infected cells, there were fibers to which both proteins (NS3 and N-protein) were found to associate.Those fibers were not coated uniformly; more gold-labeled N-protein-antibodies were found associated relative to NS3-antibodies, regardless of the gold particle size used. It is possible that antiserum produced against sucrose-gradient purified nucleocapsid protein is more antigenic than the NS3 protein used in this study, which was isolated under denaturing conditions, therefore conformational epitopes in native NS3 protein are not recognized efficiently by antibodies raised using denatured NS3 protein as antigen.
An alternative explanation is that fibrilar structures may be typical nucleocapsid, previously described for Tenuivirus, to which a few NS3 protein molecules associate. In fact, overall labeling with NS3-antibodies was less dense as compared with N-protein gold-labeled antibodies. However, low concentration of NS3-antibodies may indeed indicate that NS3 protein is expressed at lower levels in infected tissues as compared with the N protein. On the other hand, even at the same dilution, antibody titer may not comparable, which may also account for differences in the pattern of gold-labeling observed.
If N-protein and NS3 do interact, it is tempting to speculate that NS3 is the viral movement protein and associates with viral RNAor coat protein in RNPs. This would form a cell-to-cell movement complex, shuttling the viral N-protein to plasmodesmata, as in the model proposed by Lucas and Gilbertson (1994) and Citovsky and Zambrisky (1995).
However, there is still no evidence to assert that NS3 is indeed a movement protein. For instance, it is unknown whether NS3 has an RNA-binding domain or whether NS3 interacts directly with the N protein. Moreover, NS3 has not been found in plasmodesmata. Another question is whether colocalization of NS3 and N-protein in the same fibrilar structures is indicative of NS3 binding to viral RNA and formation of a single complex.
Further experiments should be carry out to assess the nature of NS3-N-protein interaction; for instance, gel retardation and band-shift assays may indicate whether NS3 has high affinity for RHBV RNA (Schoumacher et al. 1992, Thomas and Maule 1995). Subcellular localization of the NS3 may be determined by making a translational fusion with the jellyfish green fluorescent protein (GFP). Moreover, N-protein and NS3 antibodies labeled with different fluorochrome may be studied by confocal microscopy and determine whether they colocalize in the same fibrilar structures. This study may be complemented with antibodies against tubulin and actin, since movement proteins have been found associated with cytoskeleton elements (McLean et al. 1995).
Acknowlegments
The authors appreciate Reynaldo Pereira for his collaboration in the in situ localization of the NS3 protein at the Electron Microscopy Unit of the University of Costa Rica. We wish to thank to R. Hull (John Innes Centre) and F. Albertazzi for their critical review of the manuscript. This study was supported by grants from The Rockefeller Foundation and the Universidad de Costa Rica.
Resumen
El gen que codifica por la proteína no estructural NS3 del virus de la hoja blanca de arroz (RHBV) se fusionó al extremo carboxilo del gen de la glutationa-S-transferasa y se expresó en la cepa JM83 de Escherichia coli. Se obtuvieron altas concentraciones de la proteína de fusion (GST-NS3) en forma insoluble. La proteína de fusión se fraccionó en geles de SDS-PAGE, se purificó por electroelución, y se utilizó para producir anticuerpos policlonales en conejo . El antisuero producido se absorbió con extractos crudos de E. coli. Extractos crudos de plantas de arroz sanas e infectadas con el RHBV se evaluaron por Western blots detectándose una banda de peso molecular similar al estimado para la proteína NS3 (23KDa) en las plantas infectadas con el virus. Los tejidos provenientes de plantas infectadas con el RHBV se analizaron por medio de microscopia inmunoelectrónica con oro colloidal marcado con anticuerpos contra la proteína NS3 y la nucleoproteína viral N. Se observó una acumulación in situ de la proteína NS3 en el citoplasma, pero no se detectó en los cuerpos de inclusion, vacuolas o cloroplastos. Se demostró que la proteína NS3 sigue el mismo patron de distribución que el de la nucleoproteína viral N del RHBV.
References
Amann, E., J. Brosius & M. Ptashne. 1983. Vectors bearing a hybrid trp-lac promoter useful for regulated expression of cloned genes in Escherichia coli. Gene 25: 167-178. [ Links ]
Citovsky, V. & P. Zambrisky. 1995. Transport of nucleic acids through membrane channels: snaking through small holes. Ann. Rev. Microbiol. 47: 167-197. [ Links ]
de Miranda, J., M. Hernández, R. Hull & A. Espinoza. 1994. Sequence analysis of rice hoja blanca virus RNA-3. J. Gen. Virol. 75: 2127-2132 [ Links ]
de Miranda, J., R. Hull & A.M. Espinoza. 1995. Sequence of the PV2 gene of rice hoja blanca tenuivirus RNA-2. Virus Genes10: 205-209. [ Links ]
de Miranda, J., M. Muñoz, R. Wu & A.M. Espinoza. 1996. Sequence of the Rice hoja blanca tenuivirus RNA-2. Virus Genes 12: 231: 237. [ Links ]
Dean P. 1991. Affinity chromatography: a practical approach. IRL. Oxford. 213 p. [ Links ]
Espinoza, A.M., M. Hernández, R. Pereira, B. Falk & V. Medina. 1992. In situ immunogold labeling analysis of the Rice hoja blanca virus nucleoprotein and major non-capsid protein. Virology 191: 619-127. [ Links ]
Espinoza, A.M., R. Pereira, A., Macaya-Lizano, M. Hernández, M. Goulden & C. Rivera. 1993. Comparative light and electron microscopic analyses of tenuivirus major non-capsid protein (NCP) inclusion bodies in infected plants and of the NCP in vitro. Virology 195: 156-166. [ Links ]
Huiet, L., V. Klaassen, J. Tsai & B. Falk. 1991. Nucleotide sequence and RNA hybridization analyses reveal an ambisense coding strategy for maize stripe virus RNA3. Virology 182: 47-53. [ Links ]
Jennings, P. 1963. Estimating yield loss in rice caused by hoja blanca. Phytopathology 53: 492. [ Links ]
Kakutani, T., Y. Hayano, T. Hayashi & Y. Minobe. 1991. Ambisense segment 3 of rice stripe virus: the first instance of a virus containing two ambisense segments. J. Gen. Virol. 72: 465-468. [ Links ]
Laemmli, U. 1970. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature 227: 680-685. [ Links ]
Lucas, W. & R. Gilbertson. 1994. Plasmodesmata in relation to viral movement within leaf tissues. Annu. Rev. Phytopathol. 32: 387-411. [ Links ]
Marston, F. 1987. The Purification of eukaryotic polypeptides expressed in Escherichia coli. In D.M. Glover (ed.). DNA cloning: a Practical approach. Volume III. IRL, Oxford. [ Links ]
Nault, L. & E. Ammar. 1989. Leafhopper and planthopper transmission of plant viruses. Annual Review of Entomology 34: 503-529. [ Links ]
Nguyen, M., B. Ramírez, R. Goldbach & A. Haenni. 1997. Characterization of the in vitro activity of the RNA-dependent RNA polymerase associated with the ribonucleoproteins of rice hoja blanca tenuivirus. J. Gen. Virol. 71: 2621-2627. [ Links ]
Ramírez, B., G. Macaya, L. Calvert & A. Haenni. 1992. Rice hoja blanca virus genome. Characterization and expression in vitro. J. Gen. Virol. 73: 1457-1464. [ Links ]
Ramírez, B. & A. Haenni. 1994. Molecular biology of tenuiviruses, a remarkable group of plant virases J. Gen. Virol 75: 467-475. [ Links ]
Ramírez, B., I. Lozano, L. Constanino, A. Haenni & L. Calvert. 1993. Complete nucleotide sequence and coding strategy of rice hoja blanca virus RNA-4. J. Gen. Virol. 74: 2463-2468. [ Links ]
Sambrook, J., E. Fritsch & T. Maniatis. 1989. Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory, vol 3 pp. 17.3-17.42. [ Links ]
Schoumacher, F., C. Erny, A. Verna, T. Godefroy-Colburn & C. Stussi-Garaud. 1992. Nucleic acid binding properties of the alfalfa mosaic virus protein produced in yeast. Virology 188: 896-899. [ Links ]
Smith, D. & L. Corcoran. 1992. Expression and purification of glutathione-S-transferase fusio N-proteins, pp. 16.7.1-16.7.8. In F. Ausubel, R., Brent, R. Kingston, D. Moore, J. Seidman, J. Smith & K. Struhl (eds). Current Protocols in Molecular Biology, vol II. 6th. John Wiley (ed.). New York. [ Links ]
Smith, D., K. Daver, P. Board, W. Tiu, E. García & G. Mitchell. 1986. Mr 26000 antigen of Schistosoma japonicum recognized by resistant WEHI 129/J mice is a parasite Glutatione S-transferase. Proc. Natl. Acad. Sci., U.S.A. 38: 8703-8707. [ Links ]
Smith, D. & K. Johnson. 1988. Single step purification of polypeptides expressed in Escherichia coli as fusions with Glutatione S-transferase. Gene 67: 31-40. [ Links ]
Thomas, C. & A. Maule. 1995. Identification of the cauli-flower mosaic virus movement protein RNA-binding domains. Virology 206: 1145-1149. [ Links ]
Toriyama S., M.Takahashi, Y. Sano & T. Shimizu. 1994. Nucleotide sequence of RNA-1, the largest genomic segment of rice stripe virus, the prototype of the tenuiviruses. J. Gen. Virol. 75: 3569-3579. [ Links ]
Volkin, D. & A. Klibanov. 1989. Minimizing protein inactivation, pp. 1-24. In T.E. Creighton (ed.). Protein function: a practical approach. IRL, Oxford University, Oxford, England. [ Links ]

