










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.52 n.3 San José Sep. 2004
Las mutaciones inestables, nuevo reto para el consejo genético de enfermedades hereditarias
Patricia Cuenca1, 2, 3 & Fernando Morales1, 3
1 Sección de Genética Humana, Instituto de Investigaciones en Salud (INISA).
2 Escuela de Biología. Universidad de Costa Rica. 2060 San José, Costa Rica.
3 Programa Institucional de Neurociencias, Universidad de Costa Rica.
Teléfono: 224 3668; fax: 207 5130; pcuenca@cariari.ucr.ac.cr
Recibido 18-V-2003. Corregido 12-VII-2003. Aceptado 16-VIII-2004.
Abstract
Unstable mutations, new challenges for genetic counseling of inherited disorders. Unstable mutations or amplification of DNA tandem repeats sequences constitute a new kind of genetic alteration discovered in the 90´s that cause hereditary diseases. This mutation has been found inside or near important genes involved in the normal neurological function in human beings. In some cases, the presence of the amplification causes altered expression of the genes, their inactivation or the synthesis of a protein with new functions. Some common characteristics of these diseases are that they affect the central nervous system and are degenerative in nature. Most of them show genetic anticipation meaning that the severity of the manifestations increases in each generation and appear at an earlier age. In most cases, the severity of the symptoms is positively correlated with the size of the amplification. Twenty illnesses caused by this kind of mutations have been identified so far. Briefly, this work reviews the current knowledge about this topic. Rev. Biol. Trop. 52(3): 491-499. Epub 2004 Dic 15.
Key words: unstable mutations, anticipation, neurological disabilities, neurology, human genetics.
Palabras clave: mutaciones inestables, anticipación, enfermedades neurológicas, neurología, genética humana.
Las mutaciones inestables, descritas a partir del año 1991, son responsables de un número creciente de enfermedades hereditarias del sistema nervioso y multisistémicas (Fu et al. 1991, Buxton et al. 1992, Reddy y Housman 1997). La herencia de estas mutaciones constituye una nueva forma de segregación de las enfermedades hereditarias. Son segmentos de genes que cambian de tamaño en la línea germinal y como consecuencia a través de las generaciones, hasta alcanzar un tamaño que los hace patológicos. En estos casos no se pueden analizar las genealogías familiares ni estimar riesgos de recurrencia pensando en el modo mendeliano de herencia; en el cual un gen codifica un rasgo y existe una relación de tipo dominancia- recesividad entre alelos; sino que se debe conocer la cantidad exacta de repeticiones que porta cada miembro de una familia afectada, para hacer estimaciones aproximadas de los riesgos reproductivo, de recurrencia e inclusive de llegar a manifestar síntomas.
Entre las mutaciones inestables, el tipo más frecuente ocurre cuando tres nucleótidos repetidos en forma sucesiva (en "tandem") se presentan en una cantidad mayor a la normal en el gen involucrado, produciendo alteración en la función génica, la cual se puede reflejar en anormalidades del metabolismo de los ácidos ribonucleicos (ARNs), o en la ausencia o producción anormal de la proteína. Como consecuencia de esto se produce una enfermedad neurológica o multisistémica, dependiendo del gen afectado (Morell 1993). También se han encontrado mutaciones inestables en las cuales la unidad de repetición no está compuesta por tres nucleótidos, sino por cuatro, como la distrofia miotónica tipo 2, donde se repite el tetranucleótido CCTG (Liquori et al. 2001), o cinco como en el caso de la ataxia espinocerebelar tipo 10 (SCA 10), en la cual la unidad de repetición es ATTCT (Grewal et al. 2002), y hasta doce como en la epilepsia mioclónica progresiva (Potaman et al. 2003).
La cantidad de enfermedades atribuidas a este tipo de mutaciones aumenta conforme avanza la investigación sobre el fundamento molecular de las patologías neuro-degenerativas, en 1999 eran dieciocho (Cuenca y Morales 1999), ahora son veinte, por lo que es probable que en el futuro se continúen identificando otras.
Se puede intentar hacer una clasificación dependiendo de la unidad que se repite, teniendo por un lado a un grupo que son causadas por repeticiones del trinucleótido CAG (citosina, adenina, guanina). En la mayoría de ellas, la mutación se localiza en regiones codificantes de los genes, lo que causa la producción de proteínas con un segmento mayor de poliglutamina (Cuadro 1). A este grupo pertenecen la enfermedad de Huntington, las ataxias espino- erebelares (SCA) tipo 1, 2, 3, 6, 7, 12 y 17; la atrofia muscular espinobulbar o enfermedad de Kennedy y la atrofia dentatorubral-palidoluisiana. Se caracterizan por presentar pérdida neuronal progresiva en diferentes grupos de neuronas dependiendo de la enfermedad. La cadena anormal de poliglutamina confiere propiedades tóxicas a la proteína respectiva, causando como efecto final la muerte celular. La excepción en este grupo es la SCA 12, en la cual la expansión CAG no se traduce en poliglutamina, sino que se localiza en la región 5 no codificante del gen PPP2R2B que codifica para la subunidad reguladora beta de la proteína fosfatasa PP2A específica del cerebro, implicada en funciones como regulación del ciclo celular, diferenciación, funcionamiento de canales iónicos, liberación de neurotransmisores, apoptosis y otras. En este caso la expansión está en el promotor del gen y su efecto parece ser un aumento en la tasa de transcripción, alterando la función de la proteína trimérica PP2A (Holmes et al. 2001b).
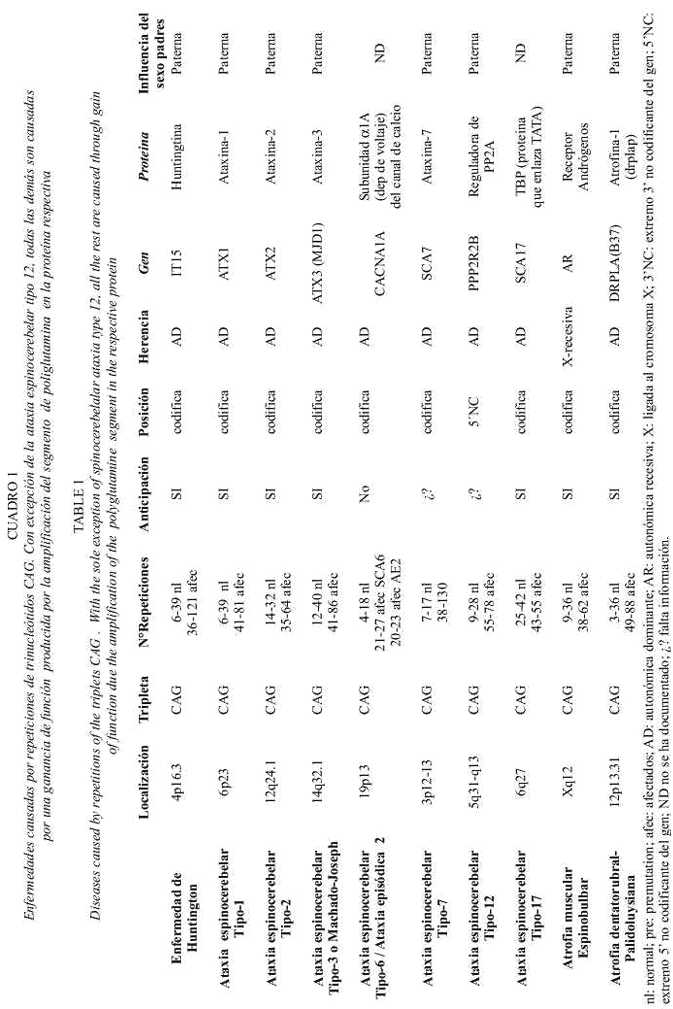
El otro grupo de estas enfermedades tienen en común el hecho de que el microsatélite amplificado es diferente de CAG y en la mayoría de los casos la mutación se localiza en una región no codificante de los genes, ya sea en un intrón, o en los extremos 3o 5 del gen respectivo. Aeste grupo pertenecen la enfermedad de Huntington tipo 2, la ataxia de Friedriech, el síndrome del cromosoma X frágil tipo A (FRAXA), el síndrome del cromosoma X frágil tipo E (FRAXE), las distrofias miotónicas (DM) tipo 1 y tipo 2, las ataxias espinocerebelares (SCA) tipo 8 y tipo 10 (Grewal et al. 2002, Potaman et al. 2003, Schöls et al. 2003). Aquí también encontramos una excepción en la enfermedad de Huntington tipo 2, en la cual la repetición CTG se localiza en el exón 1 del gen JPH3, es decir en una región codificante (Holmes et al. 2001a, Stevanin et al. 2003). Recientemente han sido descritas amplificaciones masivas del tetranucleótido CCTG dentro de un intrón en la distrofia miotónica tipo 2, al igual que en la SCA tipo 10 con el pentanucleótido ATTCT. En la epilepsia mioclónica progresiva la unidad de repetición se localiza en la punta 3´no codificante y consiste de doce nucleótidos: CCCCGCCCCGCG (Lehesjoki 2003) (cuadro 2).
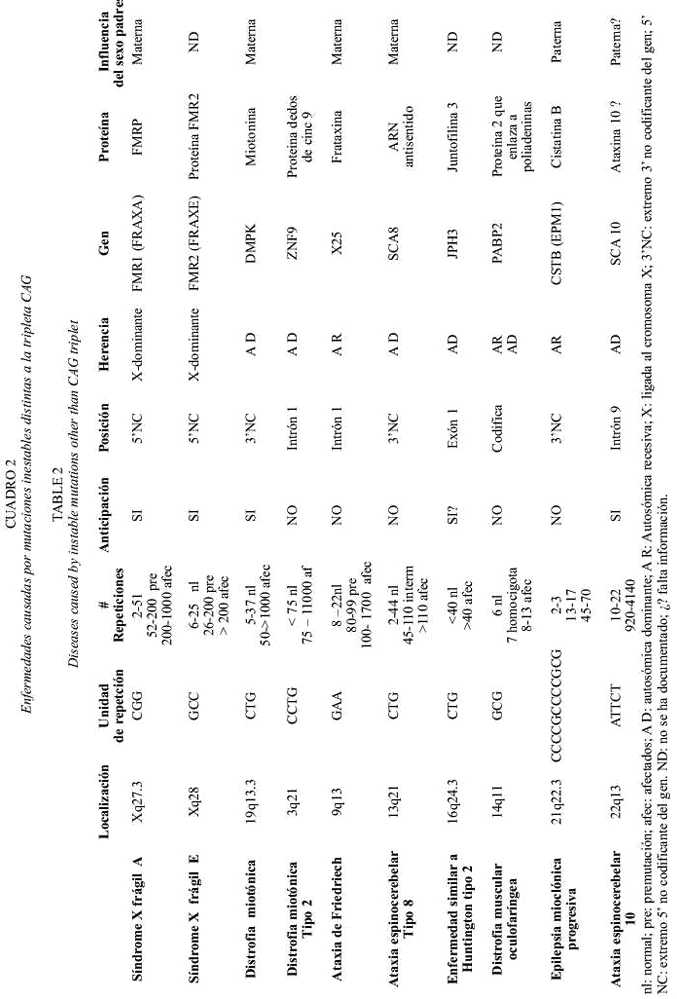
Para este grupo de enfermedades, con repeticiones diferentes a CAG, es más difícil resumir en pocas palabras el mecanismo de su patogénesis, en FRAXA la repetición causa la inactivación del gen por metilación anormal del promotor, por lo cual no se produce la proteína FMRP, que participa en el transporte de ARNs hacia las dendritas neuronales y en la regulación de la traducción del ARN propio y de otros genes (Nolin et al. 2003, Oostra y Willemsen 2003). En las distrofias miotónicas 1 y 2 el mecanismo más aceptado tiene relación con al aumento de tripletas CUG en el ARN transcrito primario del gen, causando acumulación y retención de transcritos en el núcleo de las células de pacientes con DM1 y DM2. Esta retención provoca que las proteínas de unión a los codones CUG sean secuestradas por estos ARNs, y no puedan llevar a cabo su función normal, la cual consiste en regular el procesamiento alternativo de ARNs transcritos primarios de otros genes, por lo que se ve afectada la síntesis de las proteínas alternativas normales de esos genes, es decir, se presenta una ganancia de función a nivel del ARN. El resultado de esto es la producción de dos enfermedades multisistémicas muy similares, donde están afectados el sistema muscular, nervioso, reproductor y endocrino (Liquori et al. 2001, Ranum y Day 2002, Day et al. 2003).
En las otras enfermedades de este grupo no se conoce la función exacta de las proteínas afectadas, sin embargo hay evidencia que sugiere una ganancia de función de los ARNs portadores de la expansión para la SCA8 y la SCA10, similar al que ocurre en la DM1 y la DM2 (Oostra y Willemsen 2003).
Existen también amplificaciones de la tripleta GCG que producen segmentos más largos del aminoácido alanina en genes cuyas mutaciones causan diferentes padecimientos, en el síndrome del espasmo infantil se encuentra afectado el gen ARX, OMIM 308350; en la displasia cleidocraneal es el gen CBFA1, OMIM 119600; en el síndrome del epicantus inversus tipo 2 es el gen FOXL2, OMIM 110100; en el síndrome manos-pies y genitales es el gen HOXA13, OMIM 142959; en la poli- sindactilia se trata del gen HOXD13, OMIM 142989 y en la holoprosencefalia 5 se trata del gen ZIC2, OMIM 603073 (Anónimo 2004). Sin embargo en este grupo, las amplificaciones no son la única causa de la enfermedad, sino que aparecen como una mutación más entre un grupo de mutaciones posibles de encontrar en las familias donde aparecen casos afectados (Mundlos et al. 1997, Brown et al. 1998, De Bacre et al. 2003 Sherr 2003). La expansión de segmentos de polialaninas probablemente causa agregación de las proteínas, cuyo efecto tóxico final, al igual que las agregaciones causadas por segmentos de poliglutamina, es la muerte celular (Stromme et al. 2002).
También se ha observado un segmento inestable de 24 pares de bases en el gen PRNP en algunas familias donde segrega la enfermedad de Creutzfeld-Jacob. La presencia de más de 5 repeticiones confiere susceptibilidad a esta enfermedad, OMIM 176640 (Anónimo 2004).
El descubrimiento de las mutaciones inestables motivó su búsqueda masiva en genes asociados con enfermedades de herencia compleja. De esta manera se han encontrado amplificaciones en algunos de esos genes y se ha observado hasta el momento que su presencia en ciertas poblaciones podría estar contribuyendo a aumentar el riesgo a padecer: enfermedad bipolar cuando hay amplificación en el gen SEF2-1B, OMIM 602272; autismo cuando están en el gen RELN, OMIM 600514; síndrome de Jacobsen en el gen CBL2, OMIM 147791; cáncer de ovarios en el gen hRAS1, OMIM 190020 y cáncer de próstata en el gen NCOA3, OMIM 601937 (Anónimo 2004). Sin embargo estos hallazgos no son consistentes en todos los grupos poblacionales que se han estudiado (Weitzel et al. 2000, Meira-Lima et al. 2001, Hsing et al. 2002, Zhang et al. 2002, Langdon y Armour 2003, Tamini et al. 2003).
Una de las características que presentan varias de estas enfermedades es el fenómeno de anticipación genética (ver cuadros), lo cual significa que dentro de las familias afectadas, los individuos se enferman a edades más tempranas y en forma más severa, a medida que pasan las generaciones. Este fenómeno era un misterio no resuelto que se logró entender cuando se observó que la inestabilidad inherente a estas mutaciones en la mayoría de los casos, causaba la amplificación del segmento involucrado (a veces ocurren contracciones). Ahora se sabe que las personas con síntomas más severos son también aquellas que presentan amplificaciones más grandes. La inestabilidad se presenta tanto en la línea somática como en la línea germinal, por lo tanto ocurren amplificaciones cuando se forman los gametos, en el paso de una generación a la siguiente. Así como también los individuos afectados suelen ser mosaicos somáticos para el tamaño de la amplificación como resultado de la inestabilidad somática, lo cual en algunas de estas enfermedades se ha tomado como una explicación de la variabilidad interindividual de los síntomas, porque el tamaño de la mutación varía de un tejido a otro, e inclusive intercelularmente dentro de un mismo tejido.
Los efectos fenotípicos de las amplificaciones pueden ser más complejos de lo que por ahora estamos observando y clasificando como alguna enfermedad en particular, el ejemplo más claro lo constituyen los estudios clínicos más detallados de grupos de familiares portadores de premutaciones en FRAXA (alelos dentro del rango entre 50 y 200 repeticiones); que han puesto en evidencia que la condición conocida como premutación, si bien es cierto no causa retardo mental, causa otras patologías en los adultos. Un porcentaje de las mujeres potadoras sufren problemas de aprendizaje y emocionales, y un 20% de ellas manifiestan falla ovárica prematura, lo que significa que llegan a la menopausia antes de los 40 años. Por otro lado, los varones portadores de premutaciones entre 71 y 135 repeticiones cuando llegan a la tercera edad sufren de un síndrome neurodegenerativo que se ha denominado FX-TAS (síndrome de tremor progresivo y ataxia asociado a frágil X), que puede ir acompañado de déficit en la memoria y función ejecutiva, ansiedad, parkinsonismo, neuropatía periférica y disfunción autonómica (Oostra y Willemsem 2003). Estas observaciones nos hacen pensar que dentro de la problemática que causan las mutaciones inestables son muy importantes el estudio y seguimiento de las familias completas, porque la caracterización de los efectos clínicos asociados a cada tamaño de amplificación todavía está en proceso de investigación. La afirmación muy común que se hacía hace apenas un par de años en el sentido de que se pueden sufrir amplificaciones en forma desapercibida, es decir sin efectos fenotípicos aparentes en quienes los portan, parece no ser cierta y debe investigarse para cada enfermedad.
En el Instituto de Investigaciones en Salud, donde describimos por primera vez la existencia de pacientes afectados por el síndrome del cromosoma X frágil en Costa Rica, a mediados de los ochenta (Castro y Cuenca 1987, 1996), nos abocamos a la tarea de implementar los métodos para el estudio de las mutaciones inestables a partir de 1995 (Morales et al. 2001, Cuenca et al. 2002). Nuestra principal motivación fue responder a las necesidades de muchas familias costarricenses, en las cuales súbitamente, y aparentemente sin explicación, ocurre el nacimiento o se descubre un niño con retardo mental o con la enfermedad de Steinert neonatal. Necesidades que hasta el momento no son satisfechas en forma adecuada por el sistema de salud, y que consisten básicamente en: contar con el diagnóstico exacto del padecimiento, saber que se trata de una enfermedad hereditaria y comprender todos los detalles de la transmisión con la complejidad propia de cada enfermedad, tener la posibilidad de realizarse el diagnóstico molecular para saber si se es portador de una premutación, o si se es portador asintomático de una mutación completa y conocer su riesgo reproductivo. El diagnóstico molecular también alivia a aquellos familiares que no heredaron el gen afectado y que, de no contar con esta posibilidad, hubieran renunciado a formar su propia familia.
El diagnóstico exacto de cada enfermedad confirmado por biología molecular permite también reclasificar a pacientes con diagnósticos clínicos presuntivos equivocados, a través de la búsqueda de las mutaciones correspondientes en otros genes candidatos. Esto contribuye a mejorar el manejo clínico y precisar el consejo genético (Morales et al. 2003).
Es así como hasta el momento hemos estudiado muchas familias con sospecha clínica de distrofia miotónica tipo 1, del síndrome del cromosoma X frágil, y recientemente algunas con sospecha clínica de tener la enfermedad de Huntington. (Ver publicación en este mismo número). Estas tres enfermedades son las de mayor incidencia entre las que se presentan en los cuadros 1 y 2; y por lo tanto más importantes desde el punto de vista de la salud pública. El abordaje familiar que se ofrece desde la Unidad de Consejo Genético del INISA, permitirá a mediano y largo plazo una disminución de la recurrencia de niños afectados, tal como se ha visto que ocurre en los países que practican el tamizaje activo en cascada (Murray et al. 1997). Esto significa que a partir del caso índice se buscan a los familiares para ofrecerles conocimiento actualizado sobre la enfermedad, el diagnóstico molecular y el consejo genético. En el sistema de salud tradicional, por la falta de personal capacitado en los servicios de laboratorio, y recargo de trabajo en el único Servicio de Genética con que cuenta el país, es por el momento imposible esperar que se lleve a cabo este tipo de trabajo.
Agradecimientos
A Isabel Castro Volio por la lectura crítica del manuscrito original. A mis compañeros del INISA, especialmente a Fernando Ortiz Morales, Melissa Vásquez Cerdas, Zaida Gutiérrez Castillo y Andrey Sequeira Cordero por colaborar con gran entusiasmo y mística a adaptar nuevas técnicas en el estudio de las mutaciones inestables en nuestro país. Alos neurólogos Gerardo del Valle Carazo, Roberto Brian Gago, Mauricio Sittenfeld Appel y Hubert Fernández Morales y al pediatra genetista Manuel Saborío Rocafort por depositar su confianza en nuestro laboratorio al enviarnos sus pacientes y colaborar activamente con nuestros proyectos.
Resumen
Las mutaciones inestables constituyen un tipo de alteración genética descubierta en la década de los noventa. En condiciones normales, regiones específicas de los genes están constituidas por repeticiones de una secuencia corta que puede ser de tres, cuatro, cinco o más nucleótidos; por ejemplo CAG, CGG, ATTCT, etc. Este nuevo tipo de mutación consiste en un aumento en la cantidad de éstas repeticiones, lo que causa una alteración en la expresión de dichos genes. Son inestables porque se ha observado que el tamaño de la secuencia repetida varía cuando las células se dividen por mitosis o meiosis, lo cual tiene implicaciones sobre la herencia y por consiguiente sobre el consejo genético que debe brindarse a los afectados. Estas mutaciones se han encontrado en genes importantes para la función neurológica normal del ser humano, donde pueden alterar el transporte de los ARN desde el núcleo al citoplasma, provocar la inactivación del gen o producir una proteína con funciones nuevas. La mayoría de las enfermedades causadas por estas mutaciones afectan el sistema nervioso, son degenerativas y presentan el fenómeno de anticipación genética, es decir que los afectados dentro de una familia se enfermarán más jóvenes y en forma más severa con el paso de las generaciones. La cantidad de repeticiones de la secuencia repetida presenta una correlación
negativa con la edad de manifestación, así como una correlación positiva con la severidad de la enfermedad. En este momento existen veinte padecimientos identificados que son causados exclusivamente por este tipo de mutación. Este trabajo es una breve revisión actualizada del tema.
Referencias
Brown, S.A., D. Warburton, L.Y. Brown, C.Y. Yu, E.R. Roeder, S. Stengel-Rutkowski, R.C. Hennekam & M. Muenke. 1998. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat. Genet. 20: 180-183. [ Links ]
Buxton, J., P. Shelbourne, J. Davies, C. Jones, T. Van Tongeren, C. Aslanidis, P.de Jong, G. Jansen, M. Anvert, B. Rileyu, R. Williamson & K. Johnson. 1992. Detection of an unstable fragment of DNA specific to individuals with DM. Nature 355: 547-548. [ Links ]
Castro, I. & P. Cuenca. 1987. Tamizaje del sitio frágil en el cromosoma X en una población de retardados mentales. Hallazgos preliminares. Rev. Méd. Hosp. Nal. Niños Costa Rica. 22: 11-14. [ Links ]
Castro, I. & P. Cuenca. 1996. Frecuencia del síndrome del cromosoma X frágil en la Escuela de Enseñanza Especial Fernando Centeno Guell. Acta Ped. Cost. 10: 99-106. [ Links ]
Cuenca, P. & F. Morales. 1999. Mutaciones inestables, causa de algunas enfermedades neurológicas hereditarias. Acta Méd. Cost. 41: 7-15. [ Links ]
Cuenca, P., F. Morales & I. Castro. 2002. Diagnóstico directo de la mutación que causa el síndrome del cromosoma X frágil. Experiencia en Costa Rica. Acta Méd. Cost. 44: 27-33. [ Links ]
Day, J.W., K. Ricker, J.F. Jacobsen, L.J. Rasmussen, K.A. Dick, W. Kress, C. Schneider, M.C. Koch, G.J. Beilman, A.R. Harrison, J.C. Dalton & L.P.W. Ranum. 2003. Myotonic dystrophy type 2. Molecular, diagnostic and clinical spectrum. Neurology 60: 657-664. [ Links ]
De Bacre, E., D. Beysen, C. Oley, B. Lorenz, J. Coequet, P. De Sutter, K. Devriendt, M. Dixon, M. Fellous, J.P. Fryns, A. Garza, C. Jonsrud, P.A. Koivisto, A. Krause, B.P. Leroy, F. Meire, A. Plomp L. Van Maldergem, A. De Paepe, R. Veitia, y L. Messiaen. 2003. FOXL2 and BPES : mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am. J. Hum. Genet. 72: 478-487. [ Links ]
Fu, Y.H., D.P.A Kuhl, A. Pizutti, M. Pieretti, J.S. Sutcliffe, S. Richards, A.J.M.H. Verkerk, J.J.A. Holden, R.G. Fenwick Jr., S.T. Warren, B.A. Oostra, D.L. Nelson & C.T. Caskey. 1991. Variation of the CGG repeat at the fragile X site. Results in genetic instability: Resolution of the Sherman paradox. Cell 67: 1047-1058. [ Links ]
Grewal, R.P., M. Achari, T. Matsuura, A. Durazo, E. Tayag, L. Zu, S.M. Pulst & T. Ashizawa. 2002. Clinical Features and ATTCT Repeat Expansion in Spinocerebellar Ataxia Type 10. Arch. Neurol. 59: 1285-1290. [ Links ]
Holmes, S.E., E. OHearn, A. Rosenblatt, C. Callahan, H.S. Hwang, R.G. Ingersoll-Ashworth, A. Fleisher, G. Stevanin, A. Brice, N.T. Potter, C.A. Ross & R.L. Margolis. 2001a. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat. Genet. 29: 377-378. [ Links ]
Holmes, S.E., E. OHearn, C.A. Ross & R.L. Margolis. 2001b. SCA12: An unusual mutation leads to an unusual spinocerebellar ataxia. Brain Res. Bull 56: 397-403. [ Links ]
Hsing, A.W, A.P. Chokkalingam, Y.T. Gao, G. Wu, X. Wang, J. Deng, J. Cheng, I.A. Sesterhenn, F.K. Mostofi, T. Chiang, Y.I. Chen, F.Z. Stanczyk & C. Chang. 2002. Polimorphic CAG/CAA repeat length in the AIB1/SRC-3 gene and prostate cancer risk: a population- based case-control study. Cancer Epidemiol. Biomarkers Prev. 11: 337-341. [ Links ]
Langdon, J.A. & J.A. Armour. 2003. Evolution and population genetics of the H-ras minisatellite and cancer predispotition. Hum. Mol. Genet. 15: 891-900. [ Links ]
Lehesjoki, A.E. 2003. Molecular background of progressive myoclonus epilepsy. EMBO. 14: 3473-3478. [ Links ]
Liquori, C.L., K. Ricker, M.L. Moseley, J.F. Jacobsen, W. Kress, S.L. Naylor, J.W. Day & L.P.W. Ranum. 2001. Myotonic Dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 252: 1711- 1714. [ Links ]
Meira-Lima, I.V., J. Zhao, P. Sham, A.C. Pereira, J.F. Krieger & H. Vallada. 2001. Association and linkage studies between bipolar affective disorder and the polymorphic CAG/CTG repeat loci ERDA1, SEF-1B, MAB21L and KCNN3. Mol. Psychiatry 6: 565-569. [ Links ]
Morales, F., P. Cuenca, R. Brian-Gago, M. Sittenfeld-Appel & G. del Valle-Carazo. 2001. Diagnóstico molecular de la distrofia miotónica (DM) en Costa Rica. Acta Méd. Cost. 43: 159-167. [ Links ]
Morales, F., P. Cuenca, G. del Valle, R. Brian, M. Sittenfeld, O. Montoya, T. Ashizawa, A. Rosa & Keith Johnson. 2003. Miotonía congenita: caracterización clínica de una familia costarricense afectada por la enfermedad de Thomsen. Neuroeje 17: 82-86. [ Links ]
Morell, V. 1993. The puzzle of the triple repeats. Science 260: 1422-1423. [ Links ]
Mundlos, S., F. Otto, C. Mundlos, J.B. Mulliken, A.S. Aylsworth, S. Allbright, D. Lindhout, W.G. Cole, W. Henn, J.H. Knoll, M.J. Owen, R. Mertelsmann, B.U. Zabel & B.R. Olsen. 1997. Mutation involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89: 773-779. [ Links ]
Murray, J., H. Cuckle, G. Taylor & J. Hewison,. 1997. Screening for fragile X syndrome: information needs fot health planners. J. Med. Screen. 4: 60-94. [ Links ]
Nolin, S. L., W.T. Brown, A. Glicksman, G.E. Houck Jr., A.D. Gargano, A. Sullivan, V. Biancalana, K. Broendum- Nielsen, H. Hjalgrim, E. Holinsli-Feder, F. Kooy, J. Longshore, J. Macpherson, J.L. Mandel, G. Matthijs, F. Rousseau, P. Steinbach, M.L. Vaeisaenen, H. vonKoskull & S. Sherman. 2003. Expansion of the Fragile X CGG Repeat in Females with Premutation or Intermediate Alleles. Am. J. Hum. Genet. 72: 454-464. [ Links ]
Oostra, B.A. & R. Willemsem. 2003. A fragile balance: FMR1 expression levels. Hum. Mol. Genet. 12(Review Issue 2): 249-257. [ Links ]
Potaman, V.N., J.J. Bissler, V.I. Hashem, E.A. Oussatcheva, L.L. Luda, S. Shlyakhtenko, Y.L. Lyubchenko, T. Matsuura, T. Ashizawa, M. Leefak, C.J. Benham & R. R. Sinden. 2003. Unpaired structures in SCA 10 (ATTCT)n.(AGAAT)n Repeats. J. Mol. Biol. 326: 1095-1111. [ Links ]
Ranum, L.P.W. & J.W. Day. 2002. Myotonic Dystrophy: clinical and molecular parallels between Myotonic Dystrophy tipe 1 and tipe 2. Curr. Neurol. Neurosc. Reports. 2: 465-470. [ Links ]
Reddy, P.S. & D.E. Housman. 1997. The complex patology of trinucleotide repeats. Curr. Op. Cell. Biol. 9: 364-372. [ Links ]
Sherr, E.H. 2003. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations); one gene leads to many phenotypes. Curr. Opin. Pediatr. 15: 567-571. [ Links ]
Schöls, L., I.Bauer, C. Zühlke, T. Schulte, C. Kölmel, K. Bürk, H. Topka, P. Bauer, H. Przuntek & O. Riess. 2003. Do CTG expansions at the SCA 8 locus cause ataxia? Ann. Neurol. 54: 110-115. [ Links ]
Stevanin, G., H. Fijigasaki, A.S. Lebre, A. Camuzat, C. Jeannequin, C. Dode, J. Takahashi, C. San, R. Bellance, A. Brice & A. Durr. 2003. Huntingtons disease- like phenotype due to trinucleotide repeat expansion in the TBP and JPH3 genes. Brain 126: 1599-1603. [ Links ]
Stromme, P., M.E. Mangelsdorf, M.A. Shaw, K.M. Lower, S.M. Lewis, H. Bruyere, V. Lutcherat, A.K. Gedeon, R.H. Wallace, I.E. Scheffer, G. Turner, M. Partington, S.G. Frints, J.P. Fryns, G.R. Sutherland J.C. Mullcy & J. Geez. 2002. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 30: 441-445. [ Links ]
Tamimi, R.M., S.E. Hankinson, S. Ding, V. Gagalang, G.P. Larson D. Spiegelman, G.A. Colditz, T.G. Krontiris & D.J. Hunter. 2003. The HRAS1 variable number of tandem repeats and risk of breast cancer. Cancer Epidemiol. Biomarkers Prev. 12: 1528-1530. [ Links ]
Vásquez, M., F. Morales & P. Cuenca. 2003. Aspectos genéticos y moleculares de la enfermedad de Huntington (HD). Neuroeje 17: 74-81. [ Links ]
Weitzel, J.N., S. Ding, G.P. Larson, R.A. Nelson, A. Goodman E.C. Grendys, H.G. Ball & T.G. Krontiris. 2000. The HRAS1 minisatellite locus and risk of ovarian cancer. Cancer Res. 60: 259-261. [ Links ]
Zhang, H., X. Liu, C. Zhang, E. Mudo, F. Macciardi, D.R. Grayson, A.R. Guidotti & J.J. Holden. 2002. Reelin gene alleles and susceptibility to autism spectrum disorders. Mol. Psychiatry. 7: 1012-1017. [ Links ]
Referencias de internet
Anónimo, 2004. Online Mendelian Inheritance in man. Johns Hopkins University. Baltimore. (Consultado: 14 de mayo del 2004, http:// www.ncbi.nlm.nih.gov). [ Links ]

