










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.52 n.3 San José Sep. 2004
The dystrophinopathies in Costa Rica
Jorge Azofeifa
Escuela de Biología and Instituto de Investigaciones en Salud (INISA), Universidad de Costa Rica. Ciudad Universitaria "Rodrigo Facio", 2060 San José, Costa Rica, Central America. Tel. (506) 224 3668, Fax (506) 207 5130; azofeifa@biologia.ucr.ac.cr
Received 27-XI-2003. Corrected 06-VIII-2004. Accepted 12-VIII-2004.
Abstract
A five-years long study aiming to describe the basic genetic epidemiology of the dystrophinopathies in Costa Rica recruited 31 patients with clinical symptoms of DMD/BMD at the National Childrens Hospital (HNN). This center is the obligate reference hospital of the national health system for genetic diseases, however, the geographic origin of the patients, a low percentage of deletions and a high proportion of de novo mutations found among them indicate that a significant ascertainment bias impedes a substantial scientific approach to confront and alleviate the problems posed by these severe diseases in Costa Rica. Rev. Biol. Trop. 52(3): 485- 490. Epub 2004 Dic 15.
Key words: Muscular dystrophy, Duchenne, Becker, X chromosome, carriers, deletions, haplotypes.
Palabras clave: distrofia muscular, Duchenne, Becker, cromosoma X, portadoras, deleciones, haplotipos.
The dystrophinopathies are a group of hereditary diseases mainly characterized by skeletal muscle dysfunction. They are caused by mutations at the X-linked dystrophin gene (MIM 300377) that maps to Xp21 (Murray et al. 1982). The severity of these diseases varies from myalgia and cramps to lethality at prereproductive ages resulting from a progressive muscle wasting that eventually impairs ventilation, compromising the heart and thus leading in most cases to death from cardiac failure (Emery 1993, and for a more updated review see OMIM).
The most frequent and devastating of them is Duchenne muscular dystrophy (DMD, MIM 31022) (Duchenne 1861, 1868) with an estimated incidence of 1:3 500 male births, whereas the estimated incidence of its allelic form Becker-Kiener muscular dystrophy (BMD, MIM 300376) (Becker and Kiener 1955) is 1:18 500 male births. Together they account for 40% of the muscular dystrophies (Emery 1991). Both diseases follow an X-linked recessive pattern of inheritance (Becker 1953, OMIM 2004), thus, the great majority of patients are males. However, severity and course of symptoms are different in both entities: the outcome of DMD is fatal, patients are chair-bound by the age eleven, death occurs shortly after the beginning of the second decade of life, while BMD, although physically handicapping, is milder, allowing some patients even to reproduce (for more details see Emery 1991).
One of the first genes discovered by positional cloning was the dystrophin locus (Murray et al. 1982, Koenig et al. 1987). The gene is segmented in at least 79 exons and covering 2.4 Mb, is the largest human gene known. This knowledge promptly opened the possibility to further study its fine structure and its expression patterns, as well as the nature of the disease-causing mutations. The gene shows a huge expression plasticity, which gives an idea on the many physiological roles it could play. Its full mature mRNA, in muscle for example, is 14 kb, less than 1% of the whole DNA gene extension. In addition, several full and partial transcripts are specifically expressed in different tissues because several tissue-specific promoters regulate the gene and because some of its transcripts are differentially spliced. Further details on these topics can be found in Ahn and Kunkel (1993), Tinsley et al. (1993) and OMIM.
Relation between mutation and disease
Deletion is the most common type of mutation causing dystrophinopathies. Almost two thirds of the patients present deletions ranging from small parts of the gene, segments of one exon, to large amounts involving tens of exons (Emery 1993). However, severity is not associated with the size of the deleted fragment, but with the preservation of the reading frame of the gene. Both, large and small deletions altering the sense of the message lead to DMD, whereas deletions, which, independently of their size, allow an inframe splicing of the sequence produce BMD (Gillard et al. 1989, Koenig et al. 1989, Covone et al. 1991). Five to ten percent of deleterious mutations affecting the gene are duplications, and the rest are small insertions, inversions and point mutations (Hu et al. 1990, Roberts et al. 1992, Prior et al. 1995, Baxter et al. 1997).
The cloning of the gene allowed the development of multiplex PCR protocols that make possible the fast and easy detection of deletions (Chamberlain et al. 1988). Further development of the technique increased its detection sensibility to more than 98% (Beggs et al. 1990).
The situation in Costa Rica
The theoretical knowledge on the molecular causes of dystrophinopathies and the technical improvements described, together with the fact that Costa Rica has a health profile similar to those of developed countries product of governmental sanitary polices and of a universal health system (Mohs 1982) indicated that an epidemiological genetic approach to these diseases could be attempted in the country. Therefore, a study was conducted between1998 and 2002 aiming to define the epidemiological profile of these diseases and the frequency of deletions among dystrophinopathic patients. The project was a collaboration between the University of Costa Rica (Section of Human Genetics, Institute for Health Research –INISA- and School of Medicine) and the Service of Genetics and Metabolism of the National Childrens Hospital (HNN) of the Caja Costarricense de Seguro Social (CCSS).
During these 5 years, 31 cases with clinical symptoms of dystrophinopathies, 2 BMD and 29 DMD, were detected among the patients of the HNN. The Service should be the best place where to find patients for studies like this since it is the only Genetics Service of the national health system, thus it should recruit most of the cases of the country. However, only one patient from the Province of Limon did not originate from the Central Valley of the Land (San José, Alajuela, Heredia and Cartago).
Peripheral blood was obtained from these patients and, when available, from their close female relatives (mother, sisters, maternal aunts, grandmothers). DNAs were extracted and multiplex PCRs (Chamberlain et al. 1988, 1990, Beggs et al. 1990) were performed to screen for deletions at the dystrophin gene with the modifications described elsewhere (Sancho et al. 2001). For some cases where deletions were observed, DNAs from close female relatives at risk of being carriers were tested by fluorescent dosage analysis (Azofeifa and Sancho-Fernández 2001). In addition, five dinucleotide microsatellites were used to construct haplotypes and estimate this risk for the females in a family where the index patient does not have detectable deletions (Chavarría et al. 2002).
The screening for deletions
Twelve of the 31 patients clinically diagnosed as dystrophinopatics (39%), one of them a BMD, showed deletions (Fig. 1). This is a remarkable low number since two thirds (expected 21) is a constant figure found in different studies around the world (Emery 1993) and which is in agreement with theoretical predictions based on equilibrium between selection and mutation rates (Haldane 1932, 1935). This result could be explained by a least two reasons, a biased and small sample size and/or wrong differential diagnosis of some patients since other diseases resemble the symptoms of DMD and BMD (Emery 2002).
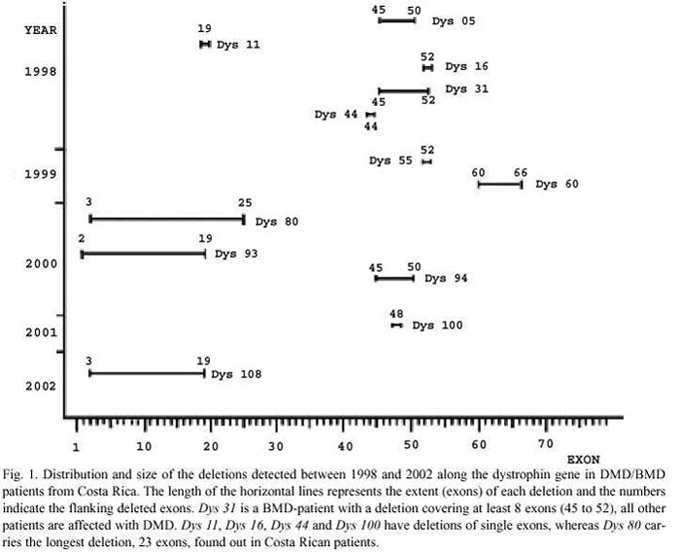
The size of the deletions found in the Costa Rican patients ranged from at least 1 to 23 exons and their distribution within the gene was as expected since they cluster around two regions known to be deletion prone, i.e. between exons 3 and 19, and between exons 44 and 52 (Fig. 1), according to different studies (Speer and Oexle 1999). However, an excess 36% (20% expected) involves deletions within the first region, and, consequently, a deficient 63% (80% expected) is found within the second region (Speer and Oexle 1999). This difference is probably due to the small number of cases with deletions found in this study.
The determination of carrier status of female relatives
The strategy to determine, or to estimate, the carrier status of at-risk female relatives of patients affected with a dystrophinopathy depends on the familial history and/or the type of mutation causing the disease. The presence of several patients in the family allow, in some cases, the identification of obligate carriers by segregation analyses. If deletions are found, a direct approach, fluorescence in situ hybridization (FISH), using as probes those segments deleted in the index patient, can detect on a chromosomal preparation the mutation. Another way to test if a female is a carrier is by fluorescence dosage analysis. If she carries the deletion affecting the index patient, she will be hemizygous for the deleted segment of the gene. Combining multiplex PCR, using primers marked with a fluorescent dye, and capillary electrophoresis, the height or the area of the peak produced by the signal of the fragment should be proportional to the quantity of molecules serving as templates, at the PCR exponential phase, for the amplification, i.e. to the dosage of the gene segment amplified. Thus, relative ratios of the area of each fragment, calculated over all the segments simultaneously amplified, should disclose if the putative carrier has an imbalance at the deleted segment when compared with a mean value obtained from normal females. Using this approach, 15 females maternally related to 6 patients (DYS 05, DYS 11, DYS 31, DYS 44, DYS 80, and DYS 94, see Fig.1) without familial history of DMD/BMD were tested (Azofeifa and Sancho-Fernández 2001). All of the ratios showed mean values similar to those of the control group indicating first, that they do not carry any deletion, and second, that the deletions documented in all 6 index-patients were originated by de novo mutations.
On the other hand, if the index patient has no identifiable deletions, an indirect method must be used to estimate risk figures. The most useful is based on the determination of haplo-types with markers around and within the dystrophin gene and a concomitant segregation analysis among all possible maternal family members, including other males (brothers, cousins). Once the haplotype of the index patient is determined its segregation pattern will indicate if the mutation was a de novo event or if it is actually segregating in the family through more female relatives. Nineteen members, 11 females and 8 males, of one family not recruited during the collaborative INISA-HNN project, where the index patient has no deletions were screened in this way (Chavarría et al. 2001). The haplotype of the patient is also carried by two of his first cousins, both healthy adults. This haplotype could be traced back to their maternal grandmother. Since there are no familial antecedents of DMD, this indicates that the mutation occurred de novo either in the germ line of the grandmother or of the mother of the patient. Anyway, as the patient has no sisters, only two brothers who inherited the X chromosome of their maternal grandfather, the mutation will no longer segregate in this family.
Summarizing, all 7 index-patients, belonging to families without previous history of dystrophinopathies, carry mutations product of de novo mutations. It is important to outline here that among the patients without deletions 7 had familial history letting us to suppose that their mothers are carriers. For several reasons no data are available on this subject for the other 18 patients recruited and their families.
If only the 7 cases analyzed are considered, there should be only one third expected to be the product of de novo mutations, in this case 2.33 patients (Emery 1993). A very plausible explanation to this finding could be that familial cases are underrepresented in this study. Among the 31 total cases studied only 9 had familial antecedents of dystrophinopathy. Given the fatal course of DMD and despite treatments offered to the patients, a natural attitude of parents would be not to bring other affected children to the physician at the clinics or hospitals, causing thus an additional ascertainment bias to the hospital-based studies (Vogel and Motulsky 1997).
Meaning of the results and perspectives
A modern approach to the dystrophinopathies in a developing country with health profile of a developed nation opened more questions than it pretended to answer. Despite the expertise of the pediatrician performing the differential diagnostics at the HNN and the installed technical capacity for molecular genetic analyses at the UCR, the study showed a fragmentary panorama of the epidemiology of these diseases: the geographic distribution of the detected cases, the small percentage of patients with deletions and the high number of de novo mutations point to a significant ascertainment bias mainly caused by lack of report and reference of patients to the HNN. This surely is a problem affecting other genetic studies in the country, a problem that could impair the further improvement of the Costa Rican health profiles. To attain such purpose, efforts must be also directed to the prevention and adequate treatment of chronic diseases, which undoubtedly have an important genetic basis. If the descriptive genetic epidemiology cannot be accurately determined the health system cannot be prepared in advance to confront the challenge.
Useful data for these studies begin with correct phenotyping, i.e. right diagnosis. This is a matter of physicians. How long will it take to have them prepared and aware of the importance of their role in this task will not only depend on medical schools but also on political commitment by the Health Ministerium and the CCSS authorities and of physicians themselves, a difficult enterprise, however feasible. In the meantime, advances in the molecular basis of genetic disease and its diagnosis will only have a minor impact on public health and on the development of significant research in Costa Rica.
Acknowledgments
Special thanks to Omar Achí, Victor Castillo, Carlos de Céspedes, Gabriela Chavarría, Daniela Macaya, André Reis, Manuel Saborío and Vanessa Sancho for their invaluable collaboration. The project was supported by the University of Costa Rica, (Project No. 742-97-253) and by the National Children Hospital (FUCODOCSA)
Resumen
Un estudio de cinco años tendiente a describir la epidemiología genética básica de las distrofinopatías en Costa Rica detectó 31 pacientes con sintomatología de DMD o de BMD en el Hospital Nacional de Niños (HNN), el centro de referencia del sistema nacional de salud para enefrmedades hereditarias, sin embargo, la distribución geográfica de los pacientes, un bajo porcentaje de deleciones y una muy elevada proporción de mutaciones de novo indican que un significante sesgo de averiguación impide el estudio científico de riguroso tendiente a disminuir el impacto de estas enfermedades en Costa Rica.
References
Ahn A.H. & L.M. Kunkel. 1993. The structural and functional diversity of dystrophin. Nat. Genet. 3: 283- 291. [ Links ]
Azofeifa J. & V.M. Sancho-Fernández. 2001. Estimación de dosis génica mediante PCR-múltiplex y electroforesis capilar flourescente en posibles portadoras de deleciones en el gen de la distrofina, Costa Rica 1998-2000. Acta Pediatr. Costarr. 15: 64-77. [ Links ]
Baxter P.S., E.L. Maltby & O. Quarrell.1997. Xp21 Muscular dystrophy due to X chromosome inversion. Neurology 49: 260. [ Links ]
Becker P.E. 1953. Dystrophia musculorum progressiva. Eine genetische und klinische Untersuchung der Muskeldystrophien. Georg Thieme, Stuttgart [ Links ]Becker P.E. & F. Kiener.1955. Eine neue X-chomosomale Muskeldystrophie. Arch. Psychiatr. Nervenkrankheiten. 193: 427-48. [ Links ]
Beggs A., M. Koenig, F. Boyce & L. Kunkel. 1990. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum. Genet. 86: 45-48. [ Links ]
Chamberlain J.S., R.A. Gibbs & J.E. Ranier. 1988. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucl. Acids Res. 16: 11141. [ Links ]
Chavarría, G., A. Reis & J. Azofeifa. 2002. Determinación indirecta, mediante marcadores de ADN, del estado de portadoras de distrofia muscular de Duchenne (DMD) en una familia costarricense. Acta Pediatr. Costarr. 16: 32-38. [ Links ]
Covone A.E., M. Lerone & G. Romeo. 1991. Genotype-phenotype correlation and germline mosaicism in DMD/BMD patients with deletions of the dystrophin gene. Hum. Genet. 87: 353-360. [ Links ]
Duchenne, G.B.A. 1861. De lElectrisation localisée et son Application à la Pathologie et à la Thérapeutique. Baillière et Fils, Paris. [ Links ]
Duchenne, G.B.A. 1868. Recherches sur la paralyse musculaire pseudohypertrophique ou paralyse mio-sclerosique. Arch. Gén. Med. 11: 5-25, 179-209, 305-21, 421-43, 552-88. [ Links ]
Emery, A.E.H. 1991. Population frequencies of inherited muscular diseases. A world survey. Neuromuscul. Disord. 1: 19-29. [ Links ]
Emery, A.E.H. 1993. Duchenne Muscular Dystrophy. Oxford University, Great Britain. 391 p. [ Links ]
Emery, A.E.H. 2002. The muscular dystrophies. Lancet 359: 687-695. [ Links ]
Haldane, J.B.S. 1932. The Causes of Evolution. Longmans Green. London. [ Links ]
Haldane, J.B.S. 1935. The rate of spontaneous mutation of a human gene. J. Genet. 31: 317-326 [ Links ]
Hu, X, P. Ray, G. Murphy, M. Thompson & R. Worton. 1990. Duplicational mutation at the Duchenne muscular dystrophy locus: its frequency, distribution, origin, and phenotype-genotype correlation. Am. J. Hum. Genet. 46: 682-695. [ Links ]
Gillard, E.F., J.S. Chamberlain, E.G. Murphy, C.L Duff, B. Smith, A.H. Burghes, M.W. Thompson, J. Sutherland, I. Oss & S.E. Bodrug. 1989. Molecular and phenotypic analysis of patients with deletions within the deletion-rich region of the Duchenne muscular dystrophy (DMD) gene. Am. J. Hum. Genet. 45: 507-520. [ Links ]
Koenig, M, E.P. Hoffman, C.J. Bertelson, A.P. Monaco, C. Feener & L.M Kunkel. 1987. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50: 509-517. [ Links ]
Mohs, E. 1982. Infectious diseases and health in Costa Rica: the development of a new paradigm. Pediatr. Infect. Dis. 1: 212-216. [ Links ]
Murray, JM, K.E. Davies, P.S. Harper, L. Meredith, C.R. Mueller & R. Williamson. 1982. Linkage relationship of a cloned DNA sequence on the short arm of the X chromosome to Duchenne muscular dystrophy. Nature 300:69-71 [ Links ]
Prior, T.W., C. Bartolo, D.K. Pearl, A.C. Papp, P.J. Zinder, M.S. Sedra, A.H. Burgh & J.R. Mendell. 1995. Spectrum of small mutations in the dystrophin coding region. Am. J. Hum. Genet. 57: 22-33. [ Links ]
Roberts, R.G., M. Bobrow & D.R. Bentley. 1992. Point mutations in the dystrophin gene. Proc. Natl. Acad. Sci. 89: 2331-2335. [ Links ]
Sancho, VM, M. Saborío, C. de Céspedes & J. Azofeifa. 2001. Tamizaje de deleciones en pacientes con distrofia muscular de Duchenne (DMD) o Becker-Kiener (BMD) mediante PCR múltiplex en Costa Rica, 1998-2000. Acta Pedatr. Costarr. 15: 78-85. [ Links ]
Speer, A. & K Oexle. 1999. Muskeldystrophien, pp. 3-30. In D. Ganten, K. Ruckpaul (eds.). Monogen bedingte Erbkrankheiten. Teil 1. Springer, Berlin. [ Links ]
Tinsley, J.M., D.J. Blake, M. Pearce, A.E. Knight, J. Kendrick-Jones & K.E. Davis. Dystrophin and related proteins. Curr. Op. Genet. Develop. 4: 82-86. [ Links ]
Vogel, F. & A. Motulsky. 1997. Human Genetics. Problems and Approaches. Springer. Berlin. [ Links ]
Internet references
OMIM (TM). 2000. Online Mendelian Inheritance in Man. Johns Hopkins University, Baltimore, MD, MIM 300377. Date of last modification 6/23/2004. (Downloaded http://www.ncbi.nlm.nih.gov/entrez/ dispomim.cgi?id=300377). [ Links ]

