










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista de Biología Tropical
On-line version ISSN 0034-7744Print version ISSN 0034-7744
Rev. biol. trop vol.52 n.3 San José Sep. 2004
Genetics of hereditary motor and sensory neuropathy and the Costa Rican contribution
Alejandro Leal
Escuela de Biología e Instituto de Investigaciones en Salud (INISA), Universidad de Costa Rica, 2060 San José, Costa Rica. Fax (506) 207-5130; aleal@cariari.ucr.ac.cr
Received 18-V-2004. Corrected 24-VIII-2004. Accepted 03-IX-2004.
Abstract
Hereditary motor and sensory neuropathy (HMSN) or Charcot-Marie-Tooth disease (CMT) is the most common hereditary illness of the peripheral nervous system. The genetics and the physiopathological aspects of the disease clarified until know, are here summarized. More than twenty genes and ten additional loci have been related with HMSN. These findings contribute to understand the metabolism of peripheral nerves and give the basis for molecular diagnostics and future therapy. Several Costa Rican families with CMT have been identified, specially with axonal forms. Two families present mutations in the myelin protein zero gene (MPZ). In addition, linkage have been found between the disease and locus 19q13.3 in an extended family, and a mutation segregating with the disease is present in a candidate gene of the critical interval. Costa Rica has several advantages for genetical studies, that can contribute importantly in the generation of knowledge in the neurogenetical field. Rev. Biol. Trop. 52(3): 475-483. Epub 2004 Dic 15.
Key words: Charcot-Marie-Tooth disease, CMT, HMSN, Genetics, Costa Rica.
Palabras clave: Enfermedad de Charcot-Marie-Tooth, CMT, HMSN, genética, Costa Rica.
Hereditary motor and sensory neuropathy (HMSN), also known as Charcot-Marie-Tooth disease (CMT), is a heterogenous neuropathical group characterized by progressive muscular weakness and atrophy of the distal muscles, often associated with mild to moderate sensory loss, foot deformities including pes cavus and pes planus, gait disturbance, and depressed deep tendon reflexes (Lupski 1998). Prevalence of CMT is estimated to be 1 in 2500-5000 people, constituting thus, the most common hereditary disease of the peripheral nervous system (Combarros et al. 1987, Holmberg 1993, Lupski et al. 1993, Skre 1974). By means of electrophysiological and morphological studies, CMT can be divided into a predominantly demyelinating type (HMSN type I) and an axonal form (HMSN type II), the latter being less frequent (Dyck et al. 1993). Primary peripheral demyelinating neuropathy is characterized by demyelination with severely reduced motor nerve conduction velocity (NCV) due to a partial or complete loss of the myelin sheath. CMT1 (autosomal dominant) and CMT4 (autosomal recessive) are forms distinguished by a slow nerve conduction, typically 10 to 30 meters per second (less than the minimal normal threshold of 38 m/s) (Table 1). On the other hand, primary axonal neuropathy results in axonal loss and normal or mildly reduced NCV (Dyck and Lambert 1968a 1968b). CMT2 (autosomal dominant) and ARCMT2 (autosomal recessive) refer to an hereditary motor and sensory neuropathy in which nerve conduction velocity is usually within the normal range, although occasionally falling, into the low normal or mildly abnormal range (35-50 m/s), but presenting reduced compound muscle action potential (CMAP) (Dyck et al. 1993). Although CMT1 and CMT2 are often clinically distinguishable, their phenotypic resemblance suggests some degree of similarity in the pathological process (Mersiyanova et al. 2000a), probably due to the close relationship between the axon and the myelin: damage in the axon leads to myelin lesion, and vice versa. This is particularly noticeable in nerve biopsies of CMT2 patients.
Around 80% of the CMT patients have a demyelinating autosomal dominant form of HMSN (Nelis et al. 1996). Clinical heterogeneity within the demyelinating forms include cases with pyramidal tract features, deafness or other signs (Dyck et al. 1993). The clinical features of CMT2 patients overlap extensively with CMT1, although, patients with CMT2 tend to be less disabled and have less sensory loss, whereas X-linked form of CMT may present an axonally pronounced form of CMT that could be confused with CMT2 (Young et al. 2001). As a matter of fact, it is not always easy to distinguish between HMSN and non-genetic neuropathy, acquired through alcoholism, vitamin 12 deficiency, thyroid disease, diabetes mellitus, HIV infection, vasculitis, leprosy, neurosyphilis, amyloid associated with chronic inflammation, occult neoplasm, or severe metal intoxication (Bird 2002). Sural nerve biopsy can help to properly diagnose CMT, but the HMSN sub-type can only be identified accurately if the molecular origin of the pathology is known.
Genetic heterogeneity and molecular cell biology of CMT
Multiple genes and loci related to this neuropathy have been identified. All of them follow Mendelian patterns of inheritance (Table 1). The high clinical and genetic heterogeneity in CMT is remarkable. Several genetic loci and genes have been related with primary demyelinating neuropathy, including altered dosage of the peripheral myelin protein 22 gene (PMP22). Seventy percent of patients with a demyelinating form carry a 1.4 Mb duplication in chromosome 17p, where PMP22 is located (Lupski et al. 1991, Patel and Lupski 1994, Timmerman et al. 1992). Additionally, CMT1 has been associated with mutations at the following genes: PMP22, connexin 32 gene or gap junction protein B1 gene (GJB1), the myelin protein zero gene (MPZ), the early growth response gene 2 (EGR2), the myotubularin- related protein 2 gene (MTMR2), the N-myc downstream-regulated protein 2 gene (NDRG1) and the periaxin gene (PRX) (Nelis et al. 2004).
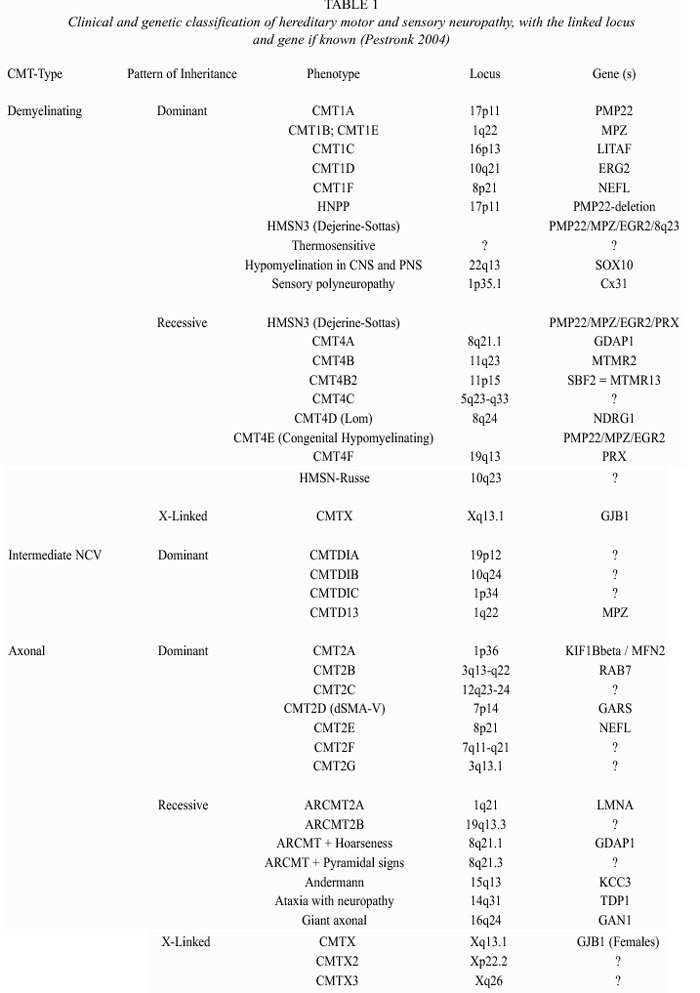
Intracellular retention in myelinating Schwann cells is probably the most common disease mechanism of PMP22 missense mutations (Naef and Suter 1999). It has been proposed that PMP22 may participate in cell proliferation, differentiation, apoptosis and cell shape definition (Jetten and Suter 2000). Delayed myelination in mice without PMP22 suggests some function in Schwann cell differentiation and the myelination process (Adlkofer et al. 1995). Both PMP22 and MPZ are components of the myelin membrane, where these two proteins form complexes (D`Urso et al. 1999). MPZ is the major component of the compact myelin sheath in the PNS (Suter 1997). Most MPZ mutations are associated with a CMT1B phenotype, but in some cases, primary axonal involvement has been found. GJB1 is a non-compact myelin protein which forms a pore unit through the myelin (Balice-Gordon et al. 1998). It has been shown, that some mutations do not disrupt the cell-cell channels, but increase the opening of hemi-channels in the non-junctional surface membrane. This increase, may damage the cells through the loss of ionic gradients and small metabolites and an increased influx of Ca2+, leading to Schwann cell damage (Abrams et al. 2002). Another gene related with demyelinating neuropathology is MTMR2, that encodes for a phosphatase that may lead to CMT4B1 through a malfunction of neural membrane recycling, membrane trafficking, endocytic or exocytotic processes, combined with altered axon-Schwann cell interaction (Berger et al. 2002). Like PMP22, NDRG1 possibly plays a role in growth arrest and cell differentiation and may be related to the complex developmental transition of the Schwann cells and Schwann cell-axonal interaction (Kalaydjieva et al. 2000). Recently, Senderek et al. (2003) found a positional candidate gene within the CMT4B2 interval, with 30% homology to MTMR2: the SET binding factor 2 gene (SBF2), encoding a member of the myotubularin family. They sequenced this gene in patients and controls and found mutations that cosegregate with the disease. Since SBF2 lacks phosphatase activity, it would act differently from MTMR2, but maybe they converge in a common pathway controlling the myelin formation in the PNS. Azzedine et al. (2003) also found mutations in this gene (that they called MTMR13) which causes demyeli-nating CMT and early-onset glaucoma. They proposed that this pseudophosphatase could be important in the development of both the peripheral nerve and the trabeculum trabecular meshwork, which permits the outflow of the aqueous humor from the eye; it is worth to remark that both of these tissues derive from the endoderm. EGR2 is a zinc-finger transcription factor that regulates the expression of myelin proteins. Mutations in the DNA-binding domain lead to dominantly inherited forms, but mutations in the NAB-protein-binding site lead to recessively inherited forms (Warner et al. 1999). Another protein, PRX, is a non-compact myelin cytoskeleton-associated protein, that links the cytoskeleton of the Schwann cell with the extra-cellular matrix (Boerkoel et al. 2002, Sherman et al. 2001). Mutations cause the CMT4F subtype or Dejerine-Sottas syndrome. On the other hand, the CMT1C syndrome is caused by mutations in a lipopolysaccharide-induced transcription factor (LITAF), that regulates the tumor necrosis factor alpha (Myokai et al. 1999, Street et al. 2003). How these mutations lead to demyelinating neuropathy is unknown. In addition, Parkinson et al. (2001) reported that mutations in the murine beta-spectrin 4 gene (Spn4) are responsible for deafness and motor neuropathy. These mutations cause alterations in the ion channel localization in the myelinated nerves, however the human homologue has not been implicated as responsible for any neuropathy.
Although the prevalence of primary peripheral axonal neuropathy is lower than demyelinating neuropathy, a considerable number of loci and genes have been related with this form. The neurofilament light chain gene (NEFL) (Mersiyanova et al. 2000b), the kinesin 1B-beta gene (KIF1B b) (Zhao et al. 2001), the mitochondrial GTPase mitofusin 2 gene (MFN2) (Zuechner et al. 2004); the gigaxonin gene (GAN1) (Bomont et al. 2000), the ganglioside-induced differentiation-associated protein 1 gene (GDAP1) (Baxter et al. 2002; Cuesta et al. 2002), the RAS-associated protein 7 gene (RAB7) (Verhoeven et al. 2003), the Lamin A/C gene (LMNA) (De Sandre-Giovannoli et al. 2002), and the glycyl tRNA synthetase gene (GARS) (Antonellis et al. 2003) have been associated with HMSN type II. Mutations in NEFL were found to be a cause of CMT2E. It encodes for one of the three neurofilament proteins in higher eukaryotes (together with medium and high neurofilaments). Brownlees et al. (2002) demonstrated that NEFL mutant neurofilaments assembly and their axonal transport in mammalian cells and neurons is disrupted. Disorganization of the neurofilament network is also present in patients with giant axonal neuropathy (affecting both CNS and PNS), due to mutations in the GAN1 gene, a member of the cytoskeletal BTB/kelch repeat family, which is involved in the organization of the actin network (Bomont et al. 2000). Zhao et al. (2001) obtained evidence that the KIF1Bb - a protein related with CMT2A (chr. 1p36) - transports synaptic vesicle precursors along the microtubules of the neurons. However, an isoform, KIF1B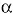 , is translated in other tissues and participates in mitochondrial transportation. Recently, Zuechner et al. (2004) found that patients with CMT2A did not present any mutation in KIF1Bb , but in the MFN2A located at 1p36 too, a gene involved in the control of the mitochondrial morphology. After this finding, several families have been found with CMT2A and mutations in MFN2A. Additionally, two research groups found mutations in the GDAP1 gene, related with both axonal and demyelinating phenotypes. GDAP1 presents a strong amino acid sequence similarity to glutathione S-transferases (GSTs) which are enzymes that play a role in cell detoxification. Cuesta et al. (2002) proposed that the mutated GDAP1 might prevent the correct catalysing S-conjugation of reduced glutathione (antioxidant), resulting in a progressive attrition of both axons and Schwann cells. Mutations in the small GTP-ase late endosomal protein RAB7 have also been related with axonal neuropathy. RAB7 belongs to the Rab family of Ras-related GTP-ases involed in the regulation of intracellular membrane trafficking and may play a role in linking vesicles and target membranes to the cytoskeleton. In fact, the Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors (Jordens et al. 2001), although it is still unknown as to how mutations in a universally expressed protein like this cause axonal CMT (Verhoeven et al. 2003). Recently, Antonellis et al. (2003) found that mutations in the glycil tRNA synthetase, an ubiquitously expressed gene, cause CMT2D and distal spinal muscular atrophy type V. Finally, the relationship between LMNA and CMT is of particular interest, since this protein was previously associated with limb-girdle muscular dystrophy type 1B, autosomal dominant Emery-Dreifuss muscular dystrophy, dilated cardiomyopathy type 1A, and autosomal dominant partial lipodystrophy. The relationship of this gene with autosomal recessive CMT type 2 (ARCMT2A) suggests that mutations in different protein domains lead to very different pathologies (De Sandre-Giovannoli et al. 2002).
, is translated in other tissues and participates in mitochondrial transportation. Recently, Zuechner et al. (2004) found that patients with CMT2A did not present any mutation in KIF1Bb , but in the MFN2A located at 1p36 too, a gene involved in the control of the mitochondrial morphology. After this finding, several families have been found with CMT2A and mutations in MFN2A. Additionally, two research groups found mutations in the GDAP1 gene, related with both axonal and demyelinating phenotypes. GDAP1 presents a strong amino acid sequence similarity to glutathione S-transferases (GSTs) which are enzymes that play a role in cell detoxification. Cuesta et al. (2002) proposed that the mutated GDAP1 might prevent the correct catalysing S-conjugation of reduced glutathione (antioxidant), resulting in a progressive attrition of both axons and Schwann cells. Mutations in the small GTP-ase late endosomal protein RAB7 have also been related with axonal neuropathy. RAB7 belongs to the Rab family of Ras-related GTP-ases involed in the regulation of intracellular membrane trafficking and may play a role in linking vesicles and target membranes to the cytoskeleton. In fact, the Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors (Jordens et al. 2001), although it is still unknown as to how mutations in a universally expressed protein like this cause axonal CMT (Verhoeven et al. 2003). Recently, Antonellis et al. (2003) found that mutations in the glycil tRNA synthetase, an ubiquitously expressed gene, cause CMT2D and distal spinal muscular atrophy type V. Finally, the relationship between LMNA and CMT is of particular interest, since this protein was previously associated with limb-girdle muscular dystrophy type 1B, autosomal dominant Emery-Dreifuss muscular dystrophy, dilated cardiomyopathy type 1A, and autosomal dominant partial lipodystrophy. The relationship of this gene with autosomal recessive CMT type 2 (ARCMT2A) suggests that mutations in different protein domains lead to very different pathologies (De Sandre-Giovannoli et al. 2002).
The costa rican contribution
In Costa Rica, epidemiological data for CMT are lacking. Nevertheless, since the public national health system is centralized, with four reference Hospitals for neuropathical cases, it is relatively easy to find patients affected by CMT and to approach their families. We have localized several families with the disease, and remarkably, most of them present an axonal polyneuropathy. Until now, our group has extensively studied five of these CMT families.
Among them, two mutations in the myelin protein zero gene have been identified as responsible for the disease in two families with both demielinating and axonal peripheral neuropathy. A Thr124Met mutation is responsible for the disease in the CR-C family with five affected individuals, all of them heterozygous for the mutation (unpublished data). In this family, affected individuals also show pupillary abnormalities and deafness. The molecular pathology of this mutation remains unclear. There may be a relationship with the role of the changed amino acid, since the variation is part of the extracellular loop close to the glycosylation site at codon 122 and the cysteine residue at codon 127, which is involved in the formation of a disulphide bridge, but it is currently unknown whether and how this mutation disrupts the normal adhesion function of the MPZ molecule (De Jonghe et al. 1999).
In family CR-D, a novel Tyr145Ser MPZ mutation was found in four affected members who also are heterozygous. In addition, two siblings were homozygous for this mutation. On neurological examination, the heterozygous parents as well as their homozygous children showed distal sensory deficits. The mother and the siblings were affected by impaired deep tendon reflexes and mild sensory ataxia; however, the homozygous individuals had an earlier age of onset and more severe distal motor weakness and pupillary abnormalities (Leal et al. 2003). It is possible to hypothesize that the Tyr145Ser MPZ mutation could cause a mild dominant form of CMT, because of a less aggressive dominant- negative effect in heterozygous patients. In the case of homozygosity, the fundamental role of P0 in adhesion and compaction would be only carried out by subnormal proteins, resulting in more severe neuropathy.
Linkage analysis was carried out in three other families affected with autosomal recessive CMT2: CR-A, CR-SR and CR-P, but in the first two families, it did not show any significant statistical relationship between the disease and a specific chromosomal region. However, after genome-wide search in the inbred family CR-P, linkage analysis of a sub-group comprising three branches of the pedigree detected linkage to the 19q13.3 region and subsequent homozygosity mapping defined shared haplotypes between marker D19S902 and D19S907 (Leal et al. 2001). A maximum two-point LOD score was obtained for marker D19S867 (Z max = 9.08) at a recombination fraction of 0.00. Clinically, the age of onset of chronic symmetric sensory-motor polyneuropathy was 28 to 42 (mean 33.8), and the electrophysiologic data clearly reflect an axonal degenerative process. Thus, phenotypically and genotypically this is a different entity from the demyelinating CMT4F mapped to 19q13.1-13.3 (Delague et al. 2000), hence the disease affecting the CR-P family constitutes an axonal, autosomal recessive CMT subtype (ARCMT2B). After the definition of the critical interval in chromosome 19q13.3 for the CR-P family, a genomic sequence contig was built up of the critical region giving some structure to the information of human draft genome sequence databases. All the genes present in this interval were analyzed, and a mutation in one gene was identified, that probably cause ARCMT2B (unpublished data). The mutation in this gene encoded protein occurred in a evolutionary domain which is highly conserved and the domain indicate close homology to a protein that plays a role in neurons, in a region with adhesion function. This domain also presents homology to a gene responsible for a neuropathy in mice. Functional analysis and screening in additional families will be performed for confirmation purposes. If this polymorphism is causative of HMSN type II, it would be a very valuable contribution in the understanding of CMT, the physiopathology of hereditary neuropathy and the function of the peripheral nerves.
Following these results, research in Costa Rican families can provide also a useful instrument for the diagnosis, prevention, and possible future therapy. Since in the near future a molecular diagnostics system for HMSN will be established in Costa Rica, both carriers test (with counseling) and contacts with interesting cases can improve. Candidate genes are going to be tested according to the phenotype; the duplication in chromosome 17, and mutations in PMP22, Cx32 and MPZ, as well as in other genes will be searched. This system will be available for neurologists and other physicians at the University of Costa Rica. Also orthopaedical and pharmacological therapies are going to be offered to patients in Costa Rica soon. All these advances announce a development in the study of neurogenetics of HMSN in Costa Rica, and also a in the medical care offered to patients. In addition, in our proteomics age, expression patterns and interactions between CMT-related proteins, and the role of these phenomena in the phenotype determination will be studied, as well as the contribution to the peripheral nerve metabolism of interactions between these proteins and the environment.
Acknowledgments
The author would like to thank Bernd Rautenstrauss, André Reis and Jorge Azofeifa for their advices and suggestions for this manuscript. Support from the Vicerrectoría de Investigación, University of Costa Rica (projects 742-98-241 and 742-A4-334) and Institute of Human Genetics, University of Erlangen-Nuremberg, is gratefully acknowledged.
Resumen
El grupo de neuropatías motoras y sensoriales hereditarias (HMSN) o enfermedad de Charcot-Marie-Tooth (CMT) es el padecimiento hereditario más común del sistema nervioso periférico. El propósito de este trabajo es resumir los aspectos genéticos y fisiopatológicos más actuales de esta enfermedad. Más de veinte genes y diez loci adicionales han sido relacionados con HMSN. Estos hallazgos han contribuido con la comprensión del metabolismo de los nervios periféricos y sirven de base para el diagnóstico molecular y el diseño de terapias. Diversas familias costarricenses con CMT han sido identificadas: dos de ellas presentan mutaciones en el gen que codifica por la mielina proteína cero (MPZ). Además, un análisis de ligamiento localizó el gen que causa una forma axonal de la enfermedad en el cromosoma 19q13.3 en una extensa familia; también se detectó en esa región una mutación que co-segrega con la enfermedad y que modifica la secuencia de un gen candidato. Costa Rica presenta numerosas ventajas para estudios genéticos, que pueden contribuir en la generación de conocimiento en el área de la neurogenética.
References
Abrams, C.K., M.V. Bennett, V.K. Verselis & T.A. Bargiello. 2002. Voltage opens unopposed gap junction hemichannels formed by a connexin 32 mutant associated with X-linked Charcot-Marie-Tooth disease. Proc. Natl. Acad. Sci. USA. 99: 3980-3984. [ Links ]
Adlkofer, K., R. Martini, A. Aguzzi, J. Zilasek, K.V. Toyka & U. Suter. 1995. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat. Genet. 11: 274-286. [ Links ]
Antonellis, A., R.E. Ellsworth, N. Sambuughin, I. Puls, A. Abel, S.Q.Lee-Lin, A. Jordanova, I. Kremensky, K. Christodoulou, L.T. Middleton, K. Sivakumar, V. Ionasescu, B. Funalot, J.M. Vance, L.G. Goldfarb, K.H. Fischbeck & E.D. Green. 2003. Glycyl tRNA Synthetase Mutations in Charcot-Marie-Tooth Disease Type 2D and Distal Spinal Muscular Atrophy Type V. Am. J. Hum. Genet. 72: 1293-1299. [ Links ]
Azzedine, H., A. Bolino, T. Taieb, N. Birouk, M. Di Duca, A. Bouhouche, S. Benamou, A. Mrabet, T. Hammadouche, T. Chkili, R. Gouider, R. Ravazzolo, A. Brice, J. Laporte & E. LeGuern. 2003. Mutations in MTMR13, a New Pseudophosphatase Homologue of MTMR2 and Sbf1, in Two Families with an Autosomal Recessive Demyelinating Form of Charcot-Marie-Tooth Disease Associated with Early-Onset Glaucoma. Am. J. Hum. Genet. 72: 1141- 1153. [ Links ]
Balice-Gordon, R.J., L.J. Bone & S.S. Scherer. 1998. Functional gap junctions in the schwann cell myelin sheath. J. Cell Biol. 142: 1095-1104. [ Links ]
Baxter, R.V., K. Ben Othmane, J.M. Rochelle, J.E. Stajich, C. Hulette, S. Dew-Knight, F. Hentati, M. Ben Hamida, S. Bel, J.E. Stenger, J.R. Gilbert, M.A. Pericak-Vance & J.M. Vance. 2002. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 30: 21-22. [ Links ]
Berger, P., P. Young & U. Suter. 2002. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenet. 4: 1-15. [ Links ]
Boerkoel, C.F., H. Takashima, C.A. Garcia, R.K. Olney, J. Johnson, K. Berry, P. Russo, S. Kennedy, A.S. Teebi, M. Scavina, L.L. Williams, P. Mancias, I.J. Butler, K. Krajewski, M. Shy & J.R. Lupski. 2002. Charcot-Marie- Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann. Neurol. 51: 190-201. [ Links ]
Bomont, P., L. Cavalier, F. Blondeau, C. Ben Hamida, S. Belal, M. Tazir, E. Demir, H. Topaloglu, R. Korinthenberg, B. Tuysuz, P. Landrieu, F. Hentati & M. Koenig. 2000. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Genet. 26: 370-374. [ Links ]
Brownlees, J., S. Ackerley, A.J. Grierson, N.J. Jacobsen, K. Shea, B.H. Anderton, P.N. Leigh, C.E. Shaw & C.C. Miller. 2002. Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Hum. Mol. Genet. 11: 2837-2844. [ Links ]
Combarros, O., J. Calleja, J.M. Polo & J. Berciano. 1987. Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol. Scand. 75: 9-12. [ Links ]
Cuesta, A., L. Pedrola, T. Sevilla, J. Garcia-Planells, M.J. Chumillas, F. Mayordomo, E. LeGuern, I. Marin, J.J. Vilchez & F. Palau. 2002. The gene encoding ganglioside- induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 30: 22-25. [ Links ]
De Jonghe, P., V. Timmerman, C. Ceuterick, E. Nelis, E. De Vriendt, A. Löfgren, A. Vercruyssen, C. Verellen, L. Van Maldergem, J. Martin & C. Van Broeckhoven. 1999. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain 122: 281-290. [ Links ]
Delague, V., C. Bareil, S. Tuffery, P. Bouvagnet, E. Chouery, S. Koussa, T. Maisonobe, J. Loiselet, A. Megarbane & M. Claustres. 2000. Mapping of a new locus for autosomal recessive demyelinating Charcot-Marie-Tooth disease to 19q13.1-13.3 in a large consanguineous Lebanese family: exclusion of MAG as a candidate gene. Am. J. Hum. Genet. 67: 236-243. [ Links ]
De Sandre-Giovannoli, A., M. Chaouch, S. Kozlov, J.M. Vallat, M. Tazir, N. Kassouri, P. Szepetowski, T. Hammadouche, A. Vandenberghe, C.L. Stewart, D. Grid & N. Levy. 2002. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 70: 726-736. [ Links ]
D`Urso, D., P. Ehrhardt & H-W. Mueller. 1999. Peripheral myelin protein 22 and protein zero: a novel association in peripheral nervous system myelin. J. Neurosci. 19: 3396-3403. [ Links ]
Dyck, P., P. Chance, R. Lebo & J. Carney. 1993. Hereditary motor and sensory neuropathies, p.p. 1094-1136. In P. Dyck, P. Thomas, J. Griffin, P. Low & J. Poduslo (eds.). Peripheral neuropathies, Saunders, Philadelphia. [ Links ]
Dyck, P.J. & E.H. Lambert. 1968a. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy: I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch. Neurol. 18: 603-618. [ Links ]
Dyck, P.J. & E.H. Lambert. 1968b. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy: II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch. Neurol. 18: 619-625. [ Links ]
Holmberg, B.H. 1993. Charcot-Marie-Tooth disease in northern Sweden: an epidemiological and clinical study. Acta Neurol. Scand. 87: 416-422. [ Links ]
Jetten, A.M. & U. Suter. 2000. The peripheral myelin protein 22 and epithelial membrane protein family. Prog Nucleic Acid. Res. Mol. Biol. 64: 97-129. [ Links ]
Jordens, I., M. Fernandez-Borja, M. Marsman, S. Dusseljee, L. Janssen, J. Calafat, H. Janssen, R. Wubbolts & J. Neefjes. 2001. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 11: 1680-1685. [ Links ]
Kalaydjieva, L., D. Gresham, R. Gooding, L. Heather, F. Baas, R. de Jonge, K. Blechschmidt, D. Angelicheva, D. Chandler, P. Worsley, A. Rosenthal, R.H. King, & P.K. Thomas. 2000. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am. J. Hum. Genet. 67: 47-58. [ Links ]
Leal, A., C. Berghoff, M. Berghoff, G. Del Valle, C. Contreras, O. Montoya, E. Hernández, R. Barrantes, A. Reis, B. Rautenstrauss & D. Heuss. 2003. Charcot-Marie-Tooth disease: A novel Tyr145Ser mutation in the Myelin Protein Zero (MPZ, P0) gene causes different phenotypes in homozygous and heterozygous carriers within one family. Neurogenet. 4: 191-197. [ Links ]
Leal, A., B. Morera, G. Del Valle, D. Heuss, C. Kayser, M. Berhoff, R. Villegas, E. Hernández, M. Méndez, H.C. Hennies, B. Neundoerfer, R. Barrantes, A. Reis & B. Rautenstrauss. 2001. A second locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to Chromosome 19q13.3. Am. J. Hum. Genet. 68: 269-274. [ Links ]
Lupski, J. 1998. Molecular genetics of peripheral neuropathies. pp. 239-256. In Martin, J. (ed.) Scientific American Molecular Neurology. New York: Scientific American Library. [ Links ]
Lupski, J.R., P.F. Chance & C.A. Garcia. 1993. Inherited primary peripheral neuropathies. Molecular genetics and clinical implications of CMT1A and HNPP. J. Am. Med. Ass. 270: 2326-2330. [ Links ]
Lupski, J.R., R.M. de Oca-Luna, S. Slaugenhaupt, L. Pentao, V. Guzzetta, B.J. Trask, O. Saucedo-Cardenas, D.F. Barker, J.M. Killian, C.A. Garcia, A. Chakravarti & P.I. Patel. 1991. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66: 219-232. [ Links ]
Mersiyanova, I.V., S.M. Ismailov, A.V. Polyakov, E.L. Dadali, V.P. Fedotov, E. Nelis, A. Lofgren, V. Timmerman, C. van Broeckhoven & O.V. Evgrafov. 2000a. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum. Mutat. 15: 340-347. [ Links ]
Mersiyanova, I.V., A.V. Perepelov, A.V. Polyakov, V.F. Sitnikov, E.L. Dadali, R.B. Oparin, A.N. Petrin & O.V. Evgrafov. 2000b. A new variant of Charcot-Marie- Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene [see comments]. Am. J. Hum. Genet. 67: 37-46. [ Links ]
Myokai, F., S. Takashiba, R. Lebo & S. Amar. 1999. A novel lipopolysaccharide-induced transcription factor regulating tumor necrosis factor alpha gene expression: molecular cloning, sequencing, characterization, and chromosomal assignment. Proc. Nat. Acad. Sci. USA 96: 4518-4523. [ Links ]
Naef, R. & U. Suter. 1999. Impaired intracellular trafficking is a common disease mechanism of PMP22 point mutations in peripheral neuropathies. Neurobiol. Dis. 6: 1-14. [ Links ]
Nelis, E., C. Van Broeckhoven, P. De Jonghe, A. Lofgren, A. Vandenberghe, P. Latour, E. Le Guern, A. Brice, M.L. Mostacciuolo, F. Schiavon, F. Palau, S. Bort, M. Upadhyaya, M. Rocchi, N. Archidiacono, P. Mandich, E. Bellone, K. Silander, M.L. Savontaus, R. Navon, H. Goldberg-Stern, X. Estivill, V. Volpini, W. Friedl & A. Gal. 1996. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur. J. Hum. Genet. 4: 25-33. [ Links ]
Parkinson, N.J., C.L. Olsson, J.L. Hallows, J. McKee-Johnson, B.P. Keogh, K. Noben-Trauth, S.G. Kujawa & B.L. Tempel. 2001. Mutant beta-spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat. Genet. 29: 61-65. [ Links ]
Patel, P.I. & J.R. Lupski. 1994. Charcot-Marie-Tooth disease: a new paradigm for the mechanism of inherited disease. T.I.G. 10: 128-132. [ Links ]
Senderek, J., C. Bergmann, S. Weber, U.P. Ketelsen, H. Schorle, S. Rudnik-Schoneborn, R. Buttner, E. Buchheim & K. Zerres. 2003. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum. Mol. Genet. 12: 349-356. [ Links ]
Sherman, D.L., C. Fabrizi, C.S. Gillespie & P.J. Brophy. 2001. Specific disruption of a schwann cell dystrophin- related protein complex in a demyelinating neuropathy. Neuron 30: 677-687. [ Links ]
Street, V.A., C.L. Bennett, J.D. Goldy, A.J. Shirk, K.A. Kleopa, B.L. Tempel, H.P. Lipe, S.S. Scherer, T.D. Bird & P.F. Chance. 2003. Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie- Tooth disease 1C. Neurology 60: 22-26. [ Links ]
Skre, H. 1974. Genetic and clinical aspects of Charcot-Marie- Tooths disease. Clin. Genet. 6: 98-118. [ Links ]
Suter, U. 1997. Myelin: keeping nerves well wrapped up. Curr. Biol. 7: R21-23. [ Links ]
Timmerman, V., E. Nelis, W. Van Hul, B.W. Nieuwenhuijsen, K.L. Chen, S. Wang, K. Ben Othman, B. Cullen, R.J. Leach & C.O. Hanemann 1992. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat. Genet. 1: 171-175. [ Links ]
Verhoeven, K., P. De Jonghe, K. Coen, N. Verpoorten, M. Auer-Grumbach, J.M. Kwon, D. FitzPatrick, E. Schmedding, E. De Vriendt, A. Jacobs, V. Van Gerwen, K. Wagner, H.P. Hartung & V. Timmerman. 2003. Mutations in the Small GTP-ase Late Endosomal Protein RAB7 Cause Charcot-Marie-Tooth Type 2B Neuropathy. Am. J. Hum. Genet. 72: 722-7. [ Links ]
Warner, L.E., C.A. Garcia & J.R. Lupski. 1999. Hereditary peripheral neuropathies: clinical forms, genetics, and molecular mechanisms. Annu. Rev. Med. 50: 263-275. [ Links ]
Young, P., K. Grote, G. Kuhlenbaumer, O. Debus, H. Kurlemann, H. Halfter, H. Funke, E.B. Ringelstein & F. Stogbauer. 2001. Mutation analysis in Chariot-Marie Tooth disease type 1: point mutations in the MPZ gene and the GJB1 gene cause comparable phenotypic heterogeneity. J. Neurol. 248: 410-415. [ Links ]
Zhao, C., J. Takita, Y. Tanaka, M. Setou, T. Nakagawa, S. Takeda, H.W. Yang, S. Terada , T. Nakata, Y. Takei, M. Saito, S. Tsuji, Y. Hayashi & N. Hirokawa. 2001. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 105: 587-597. [ Links ]
Zuechner, S., I. V. Mersiyanova, M.Muglia, N. Bissar-Tadmouri, J. Rochelle, E.L. Dadali, M. Zappia, E. Nelis, A. Patitucci, J. Senderek, Y. Parman, O. Evgrafov, P.D. Jonghe, Y. Takahashi, S. Tsuji, M.A. Pericak-Vance, A. Quattrone, E. Battaloglu, A.V. Polyakov, V. Timmerman, J.M. Schroder, J.M. Vance & E. Battologlu. 2004. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36: 449-451. [ Links ]
Internet references
Bird, T. 2002. Charcot-Marie-Tooth Neuropathy Overview. University of Washington, Seattle, USA. (Downloaded: November 12, 2003, http://www.geneclinics.org). [ Links ]
Nelis, E., M. Cruts & H. Wouters. 2004. Inherited Peripheral Neuropathies Mutation Database. University of Antwerp, Antwerp, Belgium. (Downloaded: November 12, 2003, http://www.molgen. ua.ac.be/CMTMutations/default.cfm). [ Links ]
Pestronk, A. 2004. Hereditary Motor Sensory Neuropathies Database. Washington University, St. Louis, MO, USA. (Downloaded: November 12, 2003, http://www.neuro.wustl.edu/neuromuscular/time/hm sn.html). [ Links ]

