











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkLa gama de posibilidades diagnósticas en el adulto que presenta sangrados mayores de manera espontánea es amplia.
Trastornos plaquetarios primarios, causas farmacológicas o tóxicas, enfermedad oncológica de la médula ósea, síndrome urémico, son algunas de las causas que con mayor frecuencia se encuentran en la práctica clínica. En el caso de niños con sangrado espontáneo sin causa aparente, más alteración en el tiempo de tromboplastina parcial activado (aTPT), se traslada indudablemente a la posibilidad diagnóstica de hemofilia por déficit congénito de factores de la coagulación. Sin embargo, surge un reto diagnóstico cuando un adulto sin historia de sangrado previo, se presenta con un episodio espontáneo de sangrado masivo que compromete su vida, asociado a prolongación aislada del aTPT como único dato anormal en las pruebas de coagulación. Este hallazgo sugiere una patología rara y de baja prevalencia: la hemofilia adquirida, caracterizada por el desarrollo de inhibidores adquiridos de uno o más factores de la coagulación, lo que genera depleción del factor y alteración en su función, y simula así el cuadro de una hemofilia clásica por déficit congénito de alguno de los factores.
Caso clínico
Paciente masculino de 58 años de edad, colecistectomizado hace veinte años. Antecedente de laparotomía exploratoria más lisis de adherencias tres meses previos a la presentación del cuadro clínico actual, debido a abdomen agudo obstructivo por presencia de brida; en ese momento tenía el valor de aTPT normal. No contaba con otros antecedentes patológicos, ni exposición crónica a fármacos.
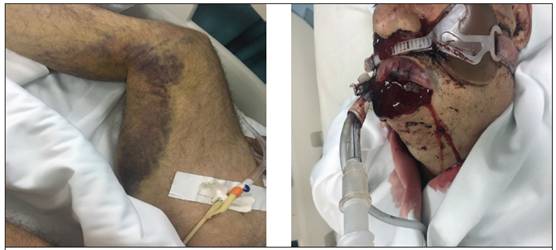
Se presenta al Servicio de Emergencias con historia de cinco días de evolución de equimosis en miembros superiores e inferiores, sin trauma previo ni causa explicable, asociado a la presencia de hematoma sublingual, de igual manera, de inicio súbito sin causa aparente, que creció con rapidez, generando dificultad respiratoria. Al ingreso al Servicio de Emergencias se documentó obstrucción de la vía aérea severa, con claudicación ventilatoria concomitante, por lo que requirió la colocación de tubo endotraqueal y ventilación mecánica asistida. Se realizó tomografía de cuello, senos paranasales y tórax, que mostraron hemosinus izquierdo, hematoma de faringe y del piso oral (Figura 1), así como del tercio proximal del esófago.
El hemograma documenta anemia normocítica normocrómica, plaquetas y serie blanca sin alteración. Las pruebas de coagulación de su ingreso indican tiempo de protrombina (TP) normal, pero aTPT prolongado con un valor inicial en 82,3 segundos, siendo este el único dato de laboratorio anormal entre las pruebas de coagulación de rutina.
Se recolectan muestras para cuantificación de factores de la coagulación e inhibidores de estos. Posterior a la recolección de muestras se transfunde plasma fresco, sin lograr reversión en los niveles prolongados del aTPT. Es admitido en la Unidad de Cuidados Intensivos, con el fin de brindar soporte ventilatorio y aféresis terapéutica con plasma fresco, ante la sospecha de enfermedad inmunomediada. El aTPT posterior a aféresis fue de 50,4 segundos. Ante la efectividad de la plasmaféresis, se sospecha hemofilia adquirida. Durante su estancia en terapia intensiva, requiere varias sesiones de aféresis, debido a que el aTPT volvía a aumentar paulatinamente” de manera aislada, junto con manifestaciones clínicas de sangrado; en ocasiones llegó a presentar niveles de más de 400 segundos (rango de referencia del laboratorio clínico, de 37 a 45 segundos). Luego se recibe reporte de cuantificación de factores de la coagulación del Centro de Investigación de Hematología y Trastornos Afines, el cual muestra presencia de inhibidor del factor VIII, con plasma normal encubado a 1 y 2 horas, a una temperatura de 37 grados centígrados, junto a un factor VIII francamente disminuido en un 1% (valor de referencia 60-150%; hemofilia grave < 1 %, hemofilia moderada 1- 5%, hemofilia leve 5-40%).
Se descartó patología de los otros factores de la vía intrínseca: IX, XI y XII, así como de la vía común. También se descartó enfermedad de von Willebrand (EvW) y presencia de inhibidores adquiridos de interferencia como anticoagulante lúpico, uso de heparina o sustancias heparinoides. Se estudió por la presencia de enfermedades autoinmunes y del colágeno, neoplasias ocultas sólidas o hematológicas, inmunodeficiencias primarias, enfermedades infecciosas, sin evidenciar la presencia de ninguna de ellas.
Se administró tratamiento inmunosupresor con metilprednisolona intravenosa 500 mg por día, por 72 horas, con posterior traslape a esteroide oral a 1mg por kilogramo de peso. Requirió de una segunda y tercera línea de tratamiento inmunosupresor para control de la enfermedad, con ciclofosfamida intravenosa 1235 mg semanales y con anticuerpo monoclonal antiCD20, rituximab, a dosis de 375 mg/m2 de superficie corporal.
El paciente evolucionó de manera satisfactoria, con control adecuado del aTPT y resolución de sus cuadros de sangrado.
Se egresó para control en Consulta Externa y continuación ambulatoria de fármacos inmunosupresores. No fue posible establecer la etiología del desarrollo del inhibidor anti-FVIII.
Discusión
Los inhibidores adquiridos de los factores de la coagulación, son anticuerpos que afectan una o varias etapas de la coagulación, manifestándose en un paciente conocido sin patologías afines a la hemostasia, con un espectro de sintomatología amplia, en el contexto de un paciente que sangra de manera inexplicable.
Esto se refleja en los exámenes de laboratorio, prolongando las pruebas de coagulación correspondientes,1,2 ya sea el aTPT, el TP, o ambos, lo que denota el defecto en vía intrínseca, extrínseca o vía común.
Los inhibidores adquiridos pueden ser del tipo específico o de interferencia; los específicos son inmunoglobulinas que se dirigen contra determinantes antigénicos o epítopos funcionales de un factor de la coagulación,1 generando modificación en depuración del factor o disfuncionalidad de este, lo que provoca manifestaciones clínicas de sangrados mayores.
Por su parte, los inhibidores adquiridos de interferencia son inmunoglobulinas que interfieren afectando la determinación de la actividad coagulante de los factores involucrados en la vía alterada.1 Ejemplo de los inhibidores de interferencia son: el anticoagulante lúpico y los heparinoides.4,5 La presencia de estos anticuerpos se ha relacionado con enfermedades autoinmunes, oncohematológicas, neoplasias sólidas, postparto, drogas farmacológicas o a causa idiopática.1-3
La literatura científica es escasa en cuanto al abordaje diagnóstico y terapéutico de esta enfermedad, debido a la infrecuencia de esta. No existen estudios aleatorizados, con evidencia suficiente, que sustenten o planteen un algoritmo de tratamiento y manejo de los pacientes afectados con esta condición patológica.
Ante la presencia de un paciente con sangrado importante asociado a un aTPT prolongado de manera aislada, como lo representa el caso expuesto, debe valorarse si existe corrección de este valor de laboratorio con plasma fresco congelado sano.
Se realiza un agregado de plasma normal al plasma enfermo, que permitirá discernir si fuese un déficit de factor o factores, en el cual el defecto corrige, o si se tratara de un efecto inhibitorio por la presencia de anticuerpos, en el cual el agregado de plasma normal no corrige la alteración.1 Una vez mezclados ambos plasmas, en caso de corrección, se debe determinar la cuantificación de los factores de la vía intrínseca FVIII, FIX, FXI, FXII. Se esperaría el descenso de un factor que traduzca su déficit. En caso de no corrección, la mezcla de ambos plasmas requiere incubación durante dos horas a 37 grados centígrados, con el fin de determinar la presencia de un inhibidor, siendo así tiempo y temperatura dependiente.1,6-8
Este método descrito originalmente para el estudio de inhibidores específicos anti-FVIII, se ha extendido a prácticamente todos los inhibidores específicos.1La diferencia es que no requiere la incubación dependiente de tiempo y de temperatura, dado que la acción de otros inhibidores es inmediata.2,6,9 No debe descartarse la presencia de inhibidor adquirido anti-FVIII sin ejecutar los ensayos de incubación de plasma normal con plasma enfermo, durante dos horas y a 37 grados centígrados.1
Debe investigarse por presencia de inhibidores de interferencia, como es el caso del anticoagulante lúpico, el cual, de igual manera que los inhibidores específicos, puede generar antiactividad de los factores de la coagulación participantes de la vía intrínseca. También debe descartarse presencia de EvW, debido a que puede generar una reducción en los niveles de FVIII. La EvW adquirida se caracteriza por la presencia de anticuerpos antifactor de von Willebrand, que inhiben su función1 y ocasionan sangrado.
En general, una vez pautado el diagnóstico, la literatura médica acuerda que el tratamiento debe dirigirse a mermar el inhibidor adquirido presente, y esto se logra a través de terapia inmunosupresora. Desde la aféresis, en aquellos casos con manifestaciones clínicas severas, partiendo del conocido principio de la terapia con aféresis en las enfermedades autoinmunes de afección sistémica grave, en conjunto con drogas inmunoterapias. Generalmente existe la necesidad de administrar dos o tres fármacos simultáneos para controlar el proceso inmunológico. En el caso presentado se necesitaron cuatro estrategias distintas para lograr control de la enfermedad, aféresis terapéutica y tres fármacos inmunosupresores/ inmunomoduladores.
Agradecimientos: al Dr. Ricardo Chinchilla Monge, microbiólogo investigador del Centro de Investigación en Hematología y Trastornos Afines

