











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkEl pioderma gangrenoso (PG) se describió por primera vez en 1916, por Brocq bajo el nombre de phagédénisme géométrique. En 1930, Brunsting et al. modificaron la descripción de la enfermedad adjudicándole el nombre actual en relación con un supuesto origen infeccioso bacteriano, y posteriormente Fulbright et al. introdujeron la hipótesis más aceptada en la actualidad que supone una respuesta inmune aberrante exagerada como probable origen de la enfermedad.1,2Su incidencia se estima en 2-3 casos por millón de habitantes al año y afecta en proporción doblemente mayor a mujeres entre la tercera y quinta década de la vida, siendo rara su presentación en la población pediátrica y en adultos mayores.2,3
La etiopatogenia de la enfermedad aún no se ha dilucidado con certeza, aunque la disfunción neutrofílica específicamente a nivel de la quimiotaxis, con mayor expresión de integrinas CR3 y CR4 junto a una inadecuada vía de señalización de estas, parece ser desencadenante de dicha entidad.2,3 Además, la evidente pérdida de la regulación de la inmunidad innata, asociación con otros procesos autoinmunes, elevación de marcadores de respuesta infamatoria sistémica como la velocidad de eritrosedimentación y la respuesta al tratamiento inmunosupresor, apoyan una etiología autoinmune de la enfermedad.1-3A continuación, con el objetivo de caracterizar la presentación clínica habitual, morfología, histología, evolución y respuesta terapéutica característica de PG ulceroso, además de resaltar la asociación de esta dermatosis con enfermedades reumatoideas, se presenta el caso clínico de una paciente portadora de AR de larga data sin previo tratamiento ni control, quien desarrolla una úlcera extensa en localización típica.
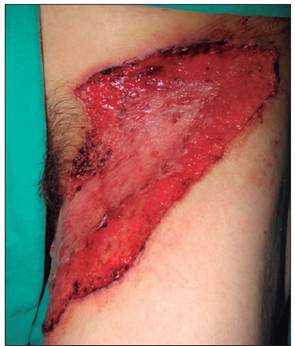
Figura 1 Lesión ulcerativa en muslo izquierdo y zona púbica, francamente dolorosa, bordes bien delimitados, sobreelevados de coloración violácea y fondo ulceroso limpio con tejido de granulación y región necrótica superior
Reporte de caso
Femenina de 38 años de edad, extabaquista (30 paquetes/año), extoxicómana (cannabis sativa y benzoilmetilecgonina), habiendo cesado dichos hábitos hace aproximadamente tres meses, con antecedente de sífilis tratada en 2015 y portadora de artritis reumatoide de más de diez años de evolución, clase funcional II (según criterios del Colegio Americano de Reumatología), sin tratamiento ni control regular debido a mala adherencia por parte de la paciente. Ninguna alergia conocida a fármacos ni antecedentes quirúrgicos o heredofamiliares de importancia. Historial ginecoobstétrico de cuatro gestas, tres partos vaginales, un aborto temprano y antecedente de prácticas sexuales de riesgo.
Se presenta con cuadro crónico de tres meses de evolución de dermatosis que inició como una pústula única sobre fondo de eritema violáceo a nivel de región púbica izquierda, la cual luego aumentó de tamaño y se ulceró de forma espontánea. La ulceración incrementó progresiva y rápidamente de tamaño durante las últimas cuatro semanas, hasta comprometer también la cara anteromedial del muslo izquierdo proximal, acompañada de intenso dolor local y escasa secreción serosanguinolenta. En ningún momento la paciente presentó síntomas constitucionales, fiebre, ni otros datos clínicos relevantes en la revisión por aparatos y sistemas.
Al examen físico se documenta paciente adelgazada, con úlcera única de 30 x 10 cm (eje longitudinal por transverso) en topografía descrita anteriormente, muy dolorosa, de borde violáceo, sobreelevado, irregular, bien delimitado, fondo limpio con tejido de granulación, sin adenopatía regional asociada (Figura.1). A nivel de codo izquierdo fenómeno de patergia y manos con cambios crónicos bilaterales simétricos, secundarios a su enfermedad reumatológica de fondo, pulgar en Z, deformidad en cuello de cisne y leve sinovitis de cuarta y quinta articulación metacarpofalángica bilateral con radiografía actual de manos típica de AR (esclerosis marginal, deformidad articular, pinzamiento del espacio articular en muñecas).
Laboratorios evidenciaron anemia leve microcítica hipocrómica, discreta trombocitosis, leucocitos y diferencial normal, reactantes de fase aguda moderadamente elevados, función hepática y renal conservadas, enzimoinmunoanálisis por virus de inmunodeficiencia humana negativo, serología por hepatitis B y C negativas, anticuerpo antinuclear positivo y factor reumatoide negativo. Se descartó etiología infecciosa mediante cultivos negativos del tejido de la lesión y la histología reveló úlcera con inflamación mixta: aguda y crónica, asociada a tejido de granulación, siendo compatible con pioderma gangrenoso. Además, mediante estudios complementarios se documentó por endoscopía digestiva alta gastritis crónica superficial y colon por enema sin ningún hallazgo patológico.
Se inició tratamiento sistémico oral con prednisona a 1mg/kg/día aunado a manejo local de la úlcera con antimicrobiano tópico e hidrocoloides, con lo cual la paciente evolucionó satisfactoriamente, logrando control adecuado y rápido del dolor, junto al cese de la extensión de la úlcera, la cual mostró epitelización en más del 75%, a los 10 días del tratamiento.
Discusión
PG corresponde a una dermatosis inflamatoria de curso crónico, recurrente, primariamente estéril, ulcerativa que se engloba en el contexto de las denominadas dermatosis neutrofílicas junto con el síndrome de Sweet, la enfermedad de Sneddon Wilkinson, el eritema elevatum diutinum, entre otros que comparten un mecanismo fisiopatogénico y hallazgos histológicos (infiltrado neutrofílico difuso y perivascular), así como la ausencia de un agente infeccioso identificable y la frecuente asociación a enfermedades sistémicas.4
Clínicamente suele presentarse tal como se manifestó en el reporte de caso de la paciente, con localización habitual en las extremidades inferiores, aunque puede también involucrar en una minoría de casos el tronco, abdomen y cara, e inicia como una pústula única, nódulos dolorosos o vesículo-pústulas múltiples que evolucionan con rapidez a úlceras con destrucción local importante, de progresión centrífuga, con bordes elevados, violáceos, bien demarcados y fondo necrótico con presencia de sangre, pus y tejido de granulación.3,5,6 De acuerdo con sus características, la úlcera se asocia a intenso dolor desproporcionado al aspecto clínico de la lesión.1
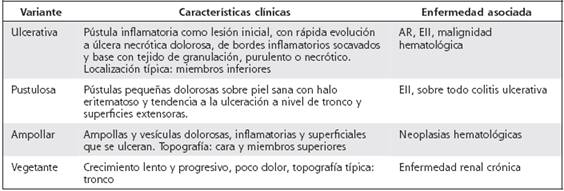
Existen cuatro variantes clínicas: ulcerosa, la más frecuente y la presentada en el caso expuesto, ampollar, pustulosa y vegetante, cada una de ellas con características específicas y en asociación a distintas enfermedades sistémicas, como se expone en el Cuadro 1. Dichas variantes pueden superponerse, aunque una suele dominar. 1,4,7,8
El PG se presenta concomitantemente con otra enfermedad sistémica en alrededor del 50-60% de los casos, sin representar una manifestación o complicación de dichas entidades cuyo curso y gravedad no tienen correlación directa con la evolución del PG.1,7Entre las patologías por lo regular asociadas se encuentran: enfermedades mieloproliferativas, lupus eritematoso sistémico, hepatitis B,C, síndrome de inmunodeficiencia adquirida, desórdenes tiroideos, AR y EII, entre otros.3,9 Esta última presente en un 30% de los casos de PG, sin embargo, solo un 2% de los pacientes portadores de dicha enfermedad manifiesta clínicamente un PG durante su evolución.7
En el caso de la AR, enfermedad de fondo de la paciente presentada, esta se ha visto asociada a PG hasta en un 37% de los casos, sobre todo en mujeres, posterior al diagnóstico de la enfermedad reumatológica, en contexto de factor reumatoide positivo y alteraciones radiográficas típicas de la enfermedad, tales como los hallazgos demostrados en radiografías de manos del caso clínico analizado.3
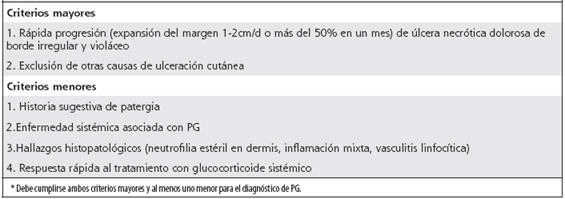
Para el diagnóstico del PG, el cual debe ser de exclusión, se han desarrollado criterios clínicos, de los cuales deben cumplirse dos de los criterios mayores y uno menor con el fin de hacer el diagnóstico certero de la enfermedad5,6,9 (Cuadro 2). El espectro de diferenciales es amplio e incluye infecciones (hongos, micobacterias, amebiasis cutánea, sífilis), malignidad (leucemia cutis y cáncer cutáneo primario), fenómenos vasculíticos (lupus, poliarteritis nodosa, granulomatosis con poliangeítis), calcifilaxis, pie diabético, úlceras por tóxicos, traumáticas y facticias.5,6
Lo histopatología del PG es inespecífica y no confirma el diagnóstico, el cual es principalmente fundamentado en las manifestaciones clínicas; sin embargo, se recomienda tomar biopsia en todos los casos con la finalidad de excluir otras enfermedades y esta no debe diferirse, pese al riesgo de patergia. Los hallazgos dependen del sitio de toma de la muestra, observándose un infiltrado linfocítico perivascular con depósito de fibrina en el borde de la úlcera, mientras que en la base predominan los neutrófilos. También el momento evolutivo de la lesión puede determinar las alteraciones histológicas, mostrando tempranamente, cambios mucho más sutiles que en las lesiones bien establecidas.1,2,3
En el caso clínico expuesto, se concluyó que la paciente presentaba un cuadro compatible con pioderma gangrenoso variante ulcerativa, asociado a AR, mediante el cumplimiento de prácticamente todos los criterios tanto mayores como menores, considerando la rápida evolución del cuadro clínico y excluyéndose etiología infecciosa ante la ausencia datos clínico-analíticos sugestivos de sepsis (no leucocitosis, adenopatías, ni fiebre), y mediante cultivos de tejido negativos. Además, la paciente no presentaba historia de trauma ni quemadura previa en la topografía de la dermatosis, no tenía antecedente de diabetes mellitus ni historial sugestivo de malignidad de fondo, como desencadenante probable, e histológicamente se descartó vasculitis y otras posibles causas del síndrome ulceroso de la paciente.
El pronóstico de la mayoría de pacientes con PG es bueno, sin embargo, a expensas de una morbilidad significativa, dado el curso usualmente crónico y recurrente de la enfermedad, que amerita en al menos la mitad de los pacientes terapia de mantenimiento para evitar recaídas.2,3,6 El fenómeno de patergia (reproducción de las lesiones en sitios expuestos a trauma), el cual estaba presente a nivel de codo izquierdo en la paciente, suele evidenciarse en un 20 - 25% de los casos, por lo que evitar traumatismos es fundamental en la prevención de nuevas lesiones.6,8
Respecto al manejo terapéutico, cabe resaltar que no existe tratamiento uniformemente efectivo y es necesario individualizar el manejo según cada paciente y la presentación clínica específica. Sin embargo, en la mayoría de casos los cuidados locales de la lesión se utilizan como coayudante a la terapia sistémica, aunque en las variantes ulcerosa leve y vegetante, pueden ser la única intervención necesaria, la cual consta del uso de apósitos hidrocoloides, control microbiano local, glucocorticoides de alta potencia, inhibidores tópicos de calcineurina y oxígeno hiperbárico para favorecer la epitelización rápida y eficaz.1,5,7 No se recomienda la debridación quirúrgica de las lesiones, dado el riesgo de patergia.3
Por otra parte, la terapia sistémica se fundamenta en fármacos inmunomosupresores e inmunomoduladores. En una primera línea de tratamiento se encuentran los esteroides sistémicos, ya sea por vía oral (prednisona) a dosis elevadas de 1mg/kg/d, o intravenoso mediante bolos de 1g/kg/d de metilprednisolona, con lo cual la mayoría de casos suele responder rápidamente, tal como lo hizo la paciente manejada con terapia esteroidea oral a la dosis recomendada, aunada a cuidados locales de la úlcera con hidrocoloides y antimicrobianos, con lo cual se logró epitelización en un 75% de la lesión, en tan solo diez días de instaurado el tratamiento.2,6
En caso de refractariedad, se han utilizado diversas terapias, entre ellas: dapsona, clofazimina, ciclosporina, tacrólimus, minociclina, micofenolato de mofetilo, y en los últimos años ha tomado auge el uso de terapia biológica con anticuerpos monoclonales antagonistas del factor de necrosis tumoral alfa, tales como el infliximab y etanercept.1,2,3
Trabajo realizado en el Servicio de Dermatología Hospital Rafael Ángel Calderón Guardia, Caja Costarricense de Seguro Social.
Abreviaturas: AR, artritis reumatoide; EII, enfermedad inflamatoria intestinal; PG, pioderma gangrenoso. eli.acon03@gmail.com

