










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.53 n.3 San José Sep. 2011
Original
Melissa Vasquez-Cerdas1,2, Domingo Campos-Ramírez1,3, Benicio Gutiérrez-Doña4, Hubert Fernández-Morales5, Fernando Morales-Montero3, Patricia Cuenca-Berger5
Lugar de realización: Instituto de Investigaciones en Salud, Universidad de Costa Rica, Hospital Nacional Psiquiátrico,
CCSS, Servicio de Neurología, Hospital "Dr. Rafael Ángel Calderón Guardia", CCSS.
1. Instituto de Investigaciones en Salud, Universidad de Costa Rica.2. Hospital Nacional Psiquiátrico
3. Programa de Investigación en Neurociencias, Universidad de Costa Rica
4. Universidad Nacional Estatal a Distancia
5. Servicio de Neurología, Hospital "Dr. Rafael Ángel Calderón Guardia", CCSS.
Abreviaturas: CAG, citosina adenina guanina; HD, enfermedad de Huntington; HNP, Hospital Nacional Psiquiátrico; PCR, reacción en cadena de la polimerasa
CorrespondenciaResumen
Objetivo: Realizar el diagnóstico molecular a personas afectadas con la enfermedad de Huntington y familiares con el 50% de riesgo, y brindarles asesoramiento genético, seguimiento y evaluación psicológica y clínica, con el fin de mejorar su calidad de vida y prevenir la ocurrencia y recurrencia de la enfermedad de Huntington.
Métodos: Diagnóstico molecular a pacientes con diagnóstico clínico, familiares asintomáticos con 50% de riesgo y pacientes con diagnóstico confuso. Se les brindó asesoramiento genético y evaluación psicológica. Los pacientes positivos que no tenían un control regular fueron referidos al neurólogo.
Resultados: El diagnóstico molecular se realizó a 64 personas (35 mujeres y 29 hombres). De estas, seis tenían diagnóstico clínico de Huntington, el cual se confirmó, y 6 tenían diagnóstico confuso; de estas últimas, cinco resultaron negativas para la enfermedad de Huntington, y una, positiva. Las restantes 52 personas correspondían a familiares en riesgo, y de estas, 17 resultaron ser portadoras. En total, 20 mujeres y 17 hombres fueron efectivamente evaluados en el nivel psicológico. Los análisis moleculares mostraron un perfil de repeticiones similar al de otras poblaciones.
Conclusión: El diagnóstico molecular es de gran ayuda, pues algunas enfermedades pueden confundirse con la de Huntington. El diagnóstico presintomático cubre satisfactoriamente las siguientes expectativas de las personas: aliviar la incertidumbre, planear el cuidado de la salud y conocer si los hijos tienen riesgo. En general, no se ha encontrado grandes diferencias entre las
personas evaluadas en el nivel psicológico, ya sea que porten un diagnóstico molecular positivo o negativo.
Descriptores: Costa Rica, asesoramiento genético, enfermedad de Huntington, diagnóstico molecular, neuropsicología.
Abstract
Aim: To perform the molecular diagnosis to affected persons and their relatives at 50% risk of Huntingtons Disease and to give genetic counselling, psychological and clinical follow-up. This will improve the clinical management of the patients that could be translated into a better quality of life for them and their families.
Methods: Molecular diagnosis for patients with clinical diagnosis of Huntington, their asymptomatic relatives at 50% risk and patients with an unclear diagnosis. The patients received genetic counselling and psychological evaluation. Positive patients for the mutation, both symptomatic and asymptomatic, who did not have a regular medical control, were referred to the neurologist for an adequate clinical management.
Results: So far, 64 individuals have been studied (35 women and 29 men) belonging to 11 unrelated and different families. Clinical diagnosis of Huntington was confirmed in six of these patients; another six patients who had an unclear diagnosis were tested but only one of them was confirmed as having Huntington. From 52 individuals at risk, 17 resulted positive for the mutation. So far, 20 women and 17 men have been evaluated at the psychological level. There are other diseases similar to Huntington, therefore, molecular diagnosis is a helpful tool in order to establish the right clinical diagnosis.
Conclusion: According to our experience, pre-symptomatic testing fully addresses the following individuals expectancies: uncertainty relief, knowledge of the risk of transmitting the disease to their children and health care planning in the coming years. No differences were found among individuals assessed at the psychological level with a positive or negative molecular diagnosis.
La enfermedad de Huntington (HD) es una enfermedad neurodegenerativa progresiva del sistema nervioso central, la cual afecta áreas determinadas del cerebro, específicamente corteza cerebral y el estriado. Las manifestaciones clínicas se caracterizan por un cuadro progresivo de movimientos anormales, rápidos e involuntarios de tipo coreico, que afectan con mayor frecuencia los miembros inferiores y la cara, además de trastornos psiquiátricos y deterioro progresivo e irreversible de las funciones cognitivas.1,2 3 La edad de manifestación de la HD es altamente variable y la incidencia en ambos sexos es similar. Por lo general, se presenta entre los 30 y 50 años de edad (HD clásica). La HD es invariablemente fatal, sin periodos de remisión, y el intervalo promedio desde el inicio de los síntomas hasta la muerte oscila entre los 15 y los 20 años, sin diferencia entre sexos.1,2,4
La enfermedad de Huntington es uno de los trastornos hereditarios del cerebro más frecuentes. Se encuentra en todos los grupos étnicos y la prevalencia está estimada entre 5 y 10 personas afectadas por cada 100 000 habitantes, en poblaciones de Europa Occidental y descendientes de estas poblaciones (poblaciones caucásicas), con igual incidencia en ambos sexos.2
La enfermedad presenta un patrón de herencia autosómico dominante con expresividad variable y penetrancia completa.5 El defecto génico consiste en una expansión del trinucleótido CAG (Citosina, Adenina, Guanina) localizado cerca del extremo 5' en el exón 1 del gen inicialmente llamado Interesting Transcript 15 (IT15), y ahora denominado gen HD, ubicado en el brazo corto del cromosoma 4 (4p16.3). Las expansiones son traducidas en un segmento de poliglutamina (poliQ) cerca del amino terminal en la proteína huntingtina (htt).6
La expansión es inestable, lo que significa que el tamaño de la secuencia repetida varía tanto en la línea germinal como en la somática. Los individuos normales poseen de 6 a 35 repeticiones CAG, mientras que los individuos con la HD poseen más de 36 repeticiones CAG.7
Los hombres y las mujeres son afectados por igual y el riesgo de transmitir el gen a la descendencia es del 50%. Todos los individuos que hereden el alelo mutado eventualmente mostrarán signos de la enfermedad, si llegan a vivir el tiempo suficiente.5
En Costa Rica no se cuenta con información epidemiológica de la HD y se desconoce exactamente su prevalencia. Los Servicios de Neurología de los hospitales nacionales no realizan estudios moleculares y en la mayoría de los casos no se cuenta con los diagnósticos clínicos confirmados por biología molecular, lo cual impide que reciban las medidas terapéuticas y de rehabilitación adecuadas, según su diagnóstico.
Al ser la HD una enfermedad neurodegenerativa, progresiva e incurable, y que se manifiesta usualmente en la adultez, en plena edad productiva, es de suma importancia conocer la población costarricense que porta la mutación. La identificación de los familiares en riesgo de desarrollarla permite brindar asesoramiento genético oportuno, seguro y confiable, basado en el diagnóstico molecular; de esta forma, las familias podrán tomar las mejores decisiones sobre su reproducción y ayudar, a largo plazo, a disminuir la ocurrencia y recurrencia de esta enfermedad. En última instancia, esto favorecerá un mejor manejo clínico de los pacientes y, por lo tanto, podrá traducirse en un incremento considerable de la calidad de vida de las familias con la HD.
Los objetivos de este estudio fueron: realizar el diagnóstico molecular a personas afectadas con la HD y sus familiares en riesgo, ofrecer asesoramiento genético y psicológico y realizar una evaluación a nivel psicológico en relación con una serie de variables, para determinar si hay diferencias entre las personas con un diagnóstico molecular positivo para la HD y uno negativo. Todo esto con el fin de mejorar la calidad de vida y prevenir la ocurrencia y recurrencia de la HD en familias costarricenses.
Materiales y métodos
Población de estudio
La población de estudio consistió de los pacientes costarricenses con un diagnóstico clínico de HD, el cual fue realizado por un neurólogo, además, los familiares de estos pacientes con riesgo de presentar la mutación que causa la HD, así como pacientes con diagnóstico incierto o confuso de HD. Los pacientes se reclutaron entre 2004 y 2009. La mayoría de los casos índices fueron localizados en el Hospital Nacional Psiquiátrico (HNP) mediante la revisión de expedientes. Los otros casos fueron referidos para estudio de HD, por neurólogos de diferentes hospitales. Tanto el protocolo del estudio como la fórmula de consentimiento informado fueron aprobados por los comités éticos científicos de la Universidad de Costa Rica y del Hospital Nacional Psiquiátrico de la Caja Costarricense de Seguro Social.
Visitas a las familias con HD e información
Una vez que se obtuvo la lista de casos de pacientes diagnosticados con la HD, se procedió a ubicar a las familias vía telefónica. Posteriormente, se visitaron sus casas de habitación para invitar a los pacientes y sus familiares a participar en el estudio. A cada paciente y sus familiares se les explicó con detalle las características clínicas y genéticas de la enfermedad y en qué consistía el proyecto. Se les informó sobre el patrón de herencia de la HD, sobre sus niveles de riesgo, el significado del estudio genético y, finalmente, se les consultó sobre la necesidad e interés de contar con los beneficios que ofrece el diagnóstico molecular de la HD: tanto el diagnóstico confirmatorio (para pacientes con diagnóstico clínico de la HD), como el diagnóstico presintomático (para individuos asintomáticos con el 50% de riesgo de padecer la HD). El objetivo final era asegurarse que la persona que iba a realizarse el diagnóstico molecular, entendiera y estuviera segura de las implicaciones de conocer el resultado. Las personas que accedieron a participar en el estudio firmaron la fórmula de consentimiento informado. Además, se entrevistó al menos a un miembro de cada familia para obtener los antecedentes familiares.
Diagnóstico molecular de la HD
Firmada la fórmula de consentimiento, a cada sujeto se le tomó una muestra de sangre periférica, de donde se obtuvo el ácido desoxirribonucleico (ADN) a partir de leucocitos mediante extracción con fenol-cloroformo, según los procedimientos usuales.9 Para determinar el número de las repeticiones CAG se realizó la reacción en cadena de la polimerasa (PCR). El protocolo de la PCR usado fue el descrito por Warner y colaboradores, 10 con algunas modificaciones.
Posteriormente, los productos de la PCR se corrieron en minigeles de agarosa al 1,5%, teñidos con bromuro de etidio, en buffer TBE 0.5X pH: 7.7 durante 1 hora, con el fin de confirmar si había producto amplificado. Los productos de la PCR se sometieron a una electroforesis sobre geles de poliacrilamida desnaturalizante al 8%, en buffer TBE 0.5X pH: 8.0, por un periodo de 2h y 50min. Para determinar el tamaño absoluto de los fragmentos, las bandas obtenidas fueron comparadas con un marcador de peso molecular. Los fragmentos fueron visualizados mediante el método de tinción con nitrato plata. Los alelos son considerados normales cuando tienen menos de 36 repeticiones, y expandidos cuando contienen más de 40 CAGs. Los alelos que tienen de 36 a 39 repeticiones presentan penetrancia incompleta.
Entrega de resultados y asesoramiento genético
Tras contar con los resultados moleculares, se contactaba al individuo vía telefónica para comunicarle que ya se tenía el resultado y para citarlo en el Instituto de Investigaciones en Salud (INISA), en procura de entregarle la información, tanto de forma oral como por escrito, siguiendo las recomendaciones del protocolo internacional. Durante las sesiones de asesoramiento se pretende traducir los conocimientos científicos en información práctica y explicar de forma clara y precisa, a cualquier persona que tenga preguntas sobre aspectos relacionados con el origen de la enfermedad, así como los mecanismos genéticos de herencia y los riesgos reproductivos.
Evaluación y apoyo psicológico
Previo a la entrega de los resultados de los análisis moleculares, psicólogos de la Escuela de Psicología y del Instituto de Investigaciones Psicológicas de la Universidad de Costa Rica, llevaron a cabo una evaluación y asesoría psicológica a los pacientes del estudio y sus familiares. La selección de las pruebas de evaluación psicológica que se aplicó fue guiada por los criterios de pertinencia y confiabilidad para muestras provenientes de la población costarricense. De esta manera, se escogieron pruebas que ya han sido utilizadas y adaptadas a la población costarricense para la investigación de la afectividad (PANAS)11, las formas de afrontamiento al estrés (Brief-COPE)12, los síntomas orgánicos asociados a estados depresivos (HOPKINS SYMPTON CHECKLIST)13, la autovaloración de la calidad de vida (Brief-WHO-QOL)14, el apoyo social (UCLA-SSI)15 y algunos factores de personalidad (MINI-MULT)16. Para comparar los pacientes con diagnóstico molecular positivo (portadores de la mutación) y los pacientes con diagnóstico molecular negativo para HD (no portadores), en relación con las variables psicológicas evaluadas y determinar si existen diferencias entre ambos grupos, se realizaron pruebas de t-student. Para efectos del artículo no se presentarán los resultados obtenidos en las pruebas de personalidad. Es importante aclarar que no se evaluaron personas que ya tenían desarrollados los síntomas asociados con la neurodegeneración avanzada.
Los pacientes debían participar de una entrevista psicológica de evaluación y asesoramiento previo a la entrega de los resultados moleculares. Para las entrevistas, fueron convocados al INISA y al Instituto de Investigaciones Psicológicas. Durante estas entrevistas los pacientes y sus familiares, si así lo deseaban, podían hacer todas las consultas y solicitar las aclaraciones que les preocupaban; de la misma manera, tenían la oportunidad de hablar de cualquier otra situación personal que les preocupara y que podría estar relacionada con el tema de la enfermedad. Además, se les hacía saber que en caso de presentar un diagnóstico molecular positivo (es decir, la mutación causal de la enfermedad), ellos serían citados nuevamente para posteriores evaluaciones similares de seguimiento.
Evaluación neurológica
Como parte de la entrega de resultados, los pacientes positivos para la HD, tanto sintomáticos como asintomáticos, que no tenían un control regular con el neurólogo, fueron referidos para seguimiento neurológico.
Resultados
Diagnóstico molecular de la HD
El diagnóstico molecular se realizó a 64 personas (pertenecientes a 11 diferentes familias costarricenses aparentemente no relacionadas): cinco fueron localizadas mediante la revisión de expedientes en el HNP, siete ingresaron al estudio por ser referidas por un neurólogo y el resto correspondían a familiares en riesgo.
De los 64 individuos, seis tenían un diagnóstico clínico de HD (tres fueron referidos por neurólogo y tres contactados mediante la revisión de expedientes en el HNP). En estos seis pacientes el diagnóstico clínico se confirmó mediante los exámenes moleculares. Otras seis personas (cinco referidas por un neurólogo y una contactada por revisión de expedientes en el HNP) tenían un diagnóstico clínico incierto (presentaban problemas en el movimiento, pero no tenían un diagnóstico clínico de HD). De estas seis personas con diagnóstico incierto, cinco resultaron negativas para la mutación y una, positiva.
Las restantes 52 personas correspondían a familiares en riesgo: hijos, hermanos, primos, sobrinos, nietos, del caso índice. Esto es así porque una vez que se localiza el caso índice o probando, se busca a los familiares para ofrecerles el diagnóstico molecular y asesoramiento genético. La mayoría de los familiares en riesgo manifestaron su interés por conocer si eran o no portadores de la mutación y la posibilidad de heredarla a sus descendientes. De los familiares en riesgo,17 resultaron ser portadores de la mutación. Al momento de este estudio, algunos de los portadores ya presentaban síntomas leves, mientras otros permanecían asintomáticos. En total participaron 35 mujeres (54,7%) y 29 hombres (45,3%).
Se logró confirmar el diagnóstico clínico de la HD en los casos índices y, por lo tanto, la presencia de la HD en seis de las familias estudiadas. En otras cuatro familias se descartó la presencia de la enfermedad y en una, el caso índice, con diagnóstico clínico de HD, ya había fallecido y los familiares analizados resultaron negativos para la mutación, sin embargo, una historia familiar positiva podría estar indicando que la HD sí está presente en la familia.
Las expansiones CAG (mutaciones) fueron observadas en 24 individuos. Las características moleculares de estos 24 individuos positivos para la mutación, se pueden observar en el Cuadro 1.
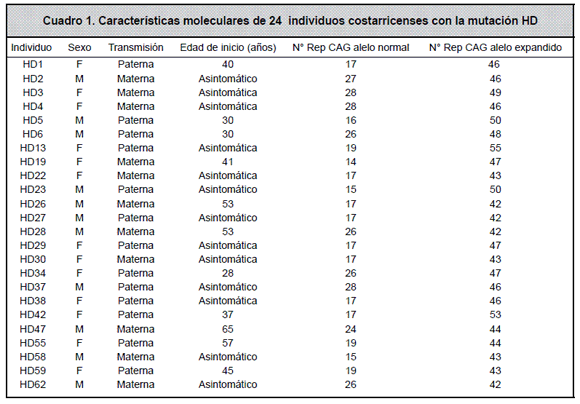
De los 24 individuos con la mutación (positivos), un 45,8% correspondían a hombres y un 54,2% a mujeres. La relación entre individuos afectados (n=11) y asintomáticos (n=13) fue 1:1.2.
Todos los 24 pacientes en los que se encontró la mutación resultaron ser heterocigotas, con un alelo normal y un alelo expandido. El número de repeticiones CAG en los alelos mutados varió de 42 a 55, con un promedio de 46 repeticiones CAG, siendo el alelo más común el de 46 repeticiones.
De los 64 participantes, 12 abandonaron en algún momento el protocolo después de haber firmado el consentimiento informado, por lo que no llegaron a recibir el resultado; los restantes sí recibieron el resultado y el respectivo asesoramiento.
Perfil psicológico de la muestra estudiada
De las 64 personas a las que se les realizó el diagnóstico molecular, solamente 37 (20 mujeres y 17 hombres) fueron efectivamente evaluadas en el nivel psicológico. El resto de las personas no se evaluaron, básicamente por dos razones: 1-abandonaron en algún momento el protocolo después de haber firmado el consentimiento informado, por lo que no llegaron a recibir el resultado, o 2-eran personas que ya tenían desarrollados los síntomas asociados con la neurodegeneración avanzada.
De estas 37 personas evaluadas, 12 presentaron un resultado positivo en el diagnóstico molecular: 7 hombres y 5 mujeres. La edad promedio de los sujetos positivos evaluados psicológicamente fue de 31,58 (D.E=13,62). Por su parte, los sujetos no afectados, 10 hombres y 15 mujeres, presentan una media de edad de 33,16 (D.E.= 14,13).
El primer análisis realizado, utilizando la escala PANAS, fue un contraste en los niveles de afectividad positiva y negativa que los sujetos reportan haber experimentado durante la última semana. Entre los sujetos positivos y los negativos para el diagnóstico molecular de HD, se ha encontrado que no se presentan diferencias estadísticamente significativas en el nivel agregado de afectividad positiva (t=1.073, g.l.=33, p=0.291); de la misma manera, no se presentan diferencias significativas en el nivel agregado de afectividad negativa (t=-1.133, g.l.=31, p=0.294). Ahora bien, cuando se toma cada una de las subescalas y se separa en cada una de las veinte emociones y sentimientos que las componen y se evalúa individualmente si existen diferencias entre los sujetos positivos y los negativos para el diagnóstico molecular en cada una de estas emociones y sentimientos, se encuentra que los sujetos con diagnóstico molecular de HD positivo presentan menores promedios en el nivel del estado de alerta (t=2.557, g.l.=35, p=0.015) y en el nivel de presencia de una actitud decidida durante la última semana a la valoración (t=2.669, g.l.=35, p=0.011); además, se halla una diferencia marginalmente significativa para el caso del nivel de irritabilidad experimentado en la última semana (t=- 1.940, gl.=34,p=0.061).
Al evaluar los estilos y las formas de afrontamiento al estrés mediante las subescalas del Brief-COPE, y contrastar los promedios entre los sujetos con diagnóstico molecular positivo y los negativos, no se encuentran diferencias estadísticamente significativas en los promedios subescalares (afrontamiento activo (t=-1.118, gl.=33,p=0.272), planificación (t=-0.033, gl.=33,p=0.973), reencuadre positivo (t=-1.511, gl.=32,p=0.141), aceptación (t=-1.513, gl.=33,p=0.140), humor (t=0.084, gl.=32,p=0.933), religión (t=-0.043, gl.=32,p=0.966), utilización de apoyo emocional (t=0.591, gl.=33,p=0.558), autodistracción (t=-0.067, gl.=31,p=0.947), negación (t=-1.372, gl.=31,p=0.180), hablar del tema (t=1.025, gl.=32,p=0.313), alcohol (t=- 1.940, gl.=34,p=0.061), renuncia (t=-1.424 gl.=30,p=0.165)).
Al consultarse por el padecimiento actual de trastornos orgánicos, se encuentra diferencias estadísticamente significativas en el promedio y tipo de padecimientos reportados entre las personas con diagnóstico positivo y negativo para HD (t=-1.054, gl.=35, p=0.299). De la misma manera, al emplear la Hopkins Symptoms Checklist para evaluar síntomas asociados con estados o rasgos depresivos, no se encuentra en la muestra diferencias estadísticamente significativas entre los portadores de la mutación para HD y los no portadores (t=0.660, gl.=24,p=.0516).
En lo que respecta a la valoración que hacen los participantes de la investigación de su calidad de vida y niveles de satisfacción con los servicios y el entorno, utilizando la Escala Abreviada de Calidad de Vida de la Organización Mundial de la Salud (WHOQOL-BREF), no se encuentra diferencias estadísticamente significativas en el contraste de medias entre los sujetos portadores de la mutación y no portadores (salud en general (t=1.406, gl.=34,p=0.169), salud física (t=-0.894, gl.=6.010,p=0.406), bienestar psicológico (t=0.813, gl.=34,p=0.422), relaciones sociales (t=0.870, gl.=33,p=0.390), ambiente y servicios (t=-0.694, gl.=7.070,p=0.510)).
Finalmente, en relación con el apoyo social percibido y las fuentes de este apoyo, se ha encontrado que las personas de la muestra con diagnóstico positivo presentan un mayor reconocimiento de haber recibido apoyo y comprensión por parte de grupos y organizaciones (t=-2,373, g.l.=10,559, p=0.038).
Es preciso aclarar que esta es una investigación en proceso, originalmente propuesta para que se realizaran varios puntos de medición a nivel psicológico: la segunda medición, al año de la primera, y solo con los casos positivos. Sin embargo, dada la cantidad de sujetos positivos y sus condiciones de ubicación geográfica, esta segunda medición se ejecuta en una muestra muy reducida, que no permite efectuar análisis estadísticos de importancia, y menos contraste de grupos.
Evaluación neurológica
En el Servicio de Neurología del Hospital Dr. Rafael A. Calderón Guardia se atiende a seis pacientes con la HD, cuyo diagnóstico se realizó por métodos moleculares. El grupo de pacientes consta de tres hombres y tres mujeres. Uno de los pacientes es totalmente asintomático, mientras que los otros muestran principalmente trastornos del movimiento, trastornos de conducta, problemas en sus estados de ánimo, ansiedad y depresión. A estos seis pacientes se les ha dado un seguimiento clínico oportuno, además de la prescripción de medicamentos en aquellos individuos que así lo necesitan. Otros pacientes son atendidos en distintos hospitales, o por neurólogos privados.
Discusión
Este trabajo ha permitido brindar un abordaje integral a los pacientes con la HD y sus familiares, incluyendo los primeros diagnósticos clínicos de esta enfermedad confirmados por estudios moleculares y los primeros diagnósticos presintomáticos que se realizan en el país, con valoración psicológica, asesoramiento genético y seguimiento clínico.
Los análisis moleculares mostraron una distribución del número de repeticiones CAG dentro de los intervalos reportados en otras poblaciones a nivel mundial (17-24). Los alelos normales más comunes a nivel mundial poseen 17 y 19 repeticiones CAG (7), y en el estudio el alelo normal más frecuente posee 17 repeticiones.
En general, los análisis genéticos son de gran utilidad, pero, hay que tener claro que, por el momento, estos diagnósticos moleculares no pueden predecir cuándo empezarán los síntomas, ni cómo será la evolución de la enfermedad. Asimismo, como se observó en cinco de los pacientes, existen muchos cuadros clínicos que presentan trastornos del movimiento y que podrían confundirse con la HD, por lo que en estos casos de duda, el diagnóstico molecular es de gran ayuda para establecer un diagnóstico clínico correcto que permita tomar las acciones del caso.
La detección de los portadores de la mutación de la HD en fases presintomáticas ahora es posible gracias a las técnicas de biología molecular, sin embargo, saber si se es portador o no de la mutación que causa la HD, puede producir consecuencias psicológicas y emocionales. Este hecho podría afectar varios aspectos de la vida del individuo, no solo el bienestar personal, sino la relación con la familia, los amigos, el trabajo y los estudios. Debido a esto, durante el proceso del diagnóstico molecular se contó con un equipo multidisciplinario que incluía psicólogos de la salud.
La intervención del equipo de psicólogos estuvo dirigida al apoyo y la asesoría psicológica emocional expresiva de los pacientes y sus familiares. El asesoramiento en el nivel psicológico se dirige, sobre todo, a escucharles y darles apoyo para que puedan expresar todas sus dudas y temores, así como sus ansiedades e inquietudes. De hecho, las personas que más han necesitado de este servicio han sido pacientes negativos para la HD, hijos de personas que resultaron positivas. Por otra parte, muchas de las manifestaciones psicológicas de ansiedad, angustia y estrés son completamente comprensibles en el marco de la situación a la que se están enfrentando; además, en la gran mayoría de los casos, los pacientes presentan una biografía personal y condiciones sociofamiliares promotoras de consecuencias emocionales y psicológicas desventajosas y tendientes a la patogenia. Algunas de estas familias se han beneficiado con referencias para que mantengan un apoyo y seguimiento psicológico en los consultorios de la Escuela de Psicología de la Universidad de Costa Rica.
Según esta experiencia y la de otros estudios, el diagnóstico presintomático cubre satisfactoriamente las siguientes expectativas de las personas que acuden a él: alivio de la incertidumbre, planeo del cuidado futuro de su salud y conocer si los hijos tienen riesgos.25 En términos generales, los individuos que se han sometido a este tipo de diagnóstico consideran que fue una buena elección.
Cuando las personas en riesgo reciben un resultado positivo para la HD (portadores de la mutación que causa la HD), mejoran su calidad de vida al reducirse la incertidumbre. Pero, la mayoría atravesará un difícil periodo de adaptación, mientras que otros pasarán por un largo periodo de estrés. A pesar de esto, la mayoría no se arrepiente de haberse realizado la prueba. En los individuos que reciben un resultado negativo (los que no son portadores de la mutación que causa la HD), si bien disminuye su angustia, a algunos les cuesta mucho adaptarse al resultado y requieren de apoyo adicional.
Esto se debe principalmente a la culpa del sobreviviente, a tener expectativas demasiado elevadas acerca de su calidad de vida después de la prueba, o a haber tomado decisiones pensando en que iban a enfermarse.26, 27
Queda claro que la enfermedad de Huntington es una patología hereditaria que tiene una connotación psicosocial importante, debido a la controversia que se establece al sopesar dos aspectos: la gravedad e irreversibilidad de los síntomas psicofísicos y su inicio generalmente tardío; lo que hace que el asesoramiento genético sea complejo. Esto, a su vez, constituye el principal incentivo a nivel mundial para las numerosas investigaciones que se llevan a cabo sobre el tema, especialmente dirigidas a lograr un tratamiento eficaz encaminado al control de los síntomas, pero mucho más esperanzadoras hacia la prevención de la neurodegeneración.28
Se espera que el abordaje familiar que se ofrece desde la Unidad de Consejo Genético del INISA, permita a mediano y largo plazos una disminución de la recurrencia de personas afectadas con la HD, tal como se ha visto que ocurre en los países que practican el tamizaje activo en cascada.29 Esto significa que a partir del caso índice se busca a los familiares para ofrecerles conocimiento actualizado sobre la enfermedad, el diagnóstico molecular y el asesoramiento genético.
Además, se ha contribuido al desarrollo de la medicina costarricense, permitiendo dar a los pacientes con la enfermedad de Huntington y sus familias, asesoramiento genético oportuno y adecuado basado en información objetiva y confiable. Se logró llenar un gran vacío en cuanto a información sobre la enfermedad, especialmente aclarando las dudas de los miembros de las familias afectadas en relación con el patrón de herencia y los riesgos de transmisión. Cuanto más completa sea la información disponible, se contribuirá a un mejor manejo de los pacientes y sus familiares. La posibilidad de conocer el diagnóstico definitivo de la HD y de interpretar estos resultados junto con la historia clínica del paciente y estudios psicológicos, se constituye en una valiosísima fuente de información para profundizar en el conocimiento de este tipo de enfermedades.
Agradecimientos y colaboradores
Al CONICIT y a la Vicerrectoría de Investigación y Vicerrectoría de Acción Social de la Universidad de Costa Rica.
Referencias
1. Landles C, Bates G. Huntingtin and the molecular pathogenesis of Huntington disease. EMBO 2004;5:958-963. [ Links ]
2. Hayden MR, Fremer B. Huntington's disease. In: Scriver CR, Sly WS, editors. The metabolic and molecular bases of inherited disease. 8a. edition. McGraw-Hill Professional, 2000:5677-5701. [ Links ]
3. Shannon KM. Movement Disorders. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, editors. Neurology in Clinical Practice. 4a edition. Butterworth-Heinemann, 2004; 2148-52. [ Links ]
4. Bates G, Harper PS, Jones L. Huntington`s disease. 3rd ed. Oxford: Oxford University. Press, 2002. [ Links ]
5. Gusella J, Young AB. Huntington`s Disease. In: Conneally M (Ed.). Molecular Basis of Neurology. Boston: Blackwell Scientific Publications; 1993. p.113-127. [ Links ]
6. The Huntington`s Disease Research Collaborative Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington`s disease chromosomes. Cell 1993;72:971-83. [ Links ]
7. Potter NT, Spector EB, Prior TW. Technical Standards and Guidelines for Huntington Disease Testing. Genet Med 2004;6(1):61-5. [ Links ]
8. Gusella JF, Wexler NS, Conneally PM, Naylor SM, Anderson MA, Tanzi RE et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 1983; 306: 234-238. [ Links ]
9. Straus WM. Preparation of genomic DNA from mammalian tissue. In Ausubel FM, R Brent, RE Kinston (Eds.). Current protocols in molecular biology. New York: Wiley; 1988.p.2.2.1-2.2.3. [ Links ]
10. Warner JP, Barron LH, Brock DJH. A new polymerase chain reaction (PCR) assay for the trinucleotide repeat that is unstable and expanded on Huntington`s disease chromosome. Mol Cell Probes 1993;7:235-39. [ Links ]
11. Watson D, Clark LA, Tellegen A. Development and validation of brief measures of positives and negatives affect: The PANAS scales. Journal of Personality and Social Psychology 1988;54:1063-1070. [ Links ]
12. Carver CS, Scheier MF, Weintraub JK. Assessing coping strategies; A theoretically based approach. Journal of Personality and Social Psycholgy 1989;56, 267-283. [ Links ]
13. Derogatis LR, Lipman RS, Rickels K, Uhlenhuth EH, Covi L. The Hopkins Symptom Checklist: a measure of primary symptom dimensions. Psychological Measurements Psychopharmacology 1974;7:79-110. [ Links ]
14. Ferrans CE, Powers MJ. Quality of Life Index: Development and Psychometric Properties. Advances in Nursery Sciences 1985; 8: 15-24. [ Links ]
15. Scharzer R, Dunkel-Schetter Ch, Kemeny M. The Multidimensional Nature of Received Social Support in Gay Men at Risk of HIV Infection and AIDS. American Journal of Community Psychology 1994;22: 319-330. [ Links ]
16. Kincannon J. Prediction of the Standart MMPI Scale Scores from 71 Items: The Mni-Mult. Journal of Consulting and Clinical Psychology 1968;32: 319-325. [ Links ]
17. Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington´s disease. Nat Genet 1993; 4: 398-403. [ Links ]
18. Whitefield JE, Williams L, Snow K, Dixon J, Winship I, Stapleton PM et al. Molecular analysis of the Huntington´s disease gene in New Zealand. N Z Med J 1996;109(1015):27-30. [ Links ]
19. Sánchez A, Castellvi-Bel S, Mila M, Genis D, Calopa M, Jiménez D et al. Huntington´s disease: confirmation of diagnosis andpresymptomatic testing in spanish families by genetic analysis. J Neurol Neurosurg Psychiatry 1996;61: 625-7. [ Links ]
20. Alonso ME, Yescas P, Cisneros B, Martínez C, Silva G, Ochoa A et al. Analysis of the (CAG)n repeat causing Huntington´s disease in a Mexican population. Clin Genet 1997;51:225-30. [ Links ]
21. Lima e Silva T, Guerra H, Bertuzzo C, Lopes I. Molecular diagnosis of Huntington disease in Brazilian patients. Arq Neuropsiquiatr 2000;58: 11-17. [ Links ]
22. Saleem Q, Roy S, Murgood U, Saxena R, Verma IC, Anand A et al. Molecular analysis of Huntington´s disease and linked polymorphisms in the Indian population. Acta Neurol Scand 2003;108:281-86. [ Links ]
23. Akbas F, Erginel-Unaltuna N. DNA testing for Huntington disease in the Turkish population. Eur Neurol 2003;50: 20-4. [ Links ]
24. The U.S.–Venezuela Collaborative Research Project. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington`s disease age of onset. Proc Nat Acad Sci 2004;101: 498-503. [ Links ]
25. Rasmussen A, Alonso E. El diagnóstico predictivo genético y sus implicaciones. Salud Mental 2002;25: 9-13. [ Links ]
26. Dudokdewit AC, Tibben A, Duivenvoorden HJ, Niermeijer MF, Passchier J, Trijsburg RW. Distress in individuals facing predictive DNA testing for autosomal dominant late-onset disorders: comparing questionnaire results with in-depth interviews. Am J Med Genet 1998;75:62-74. [ Links ]
27. Hayden MR, Bloch M, Wiggins S. Psychological effects of predictive testing for Huntington`s disease. Adv Neurol 1995;65:201-210. [ Links ]
28. Encinosa, G. Corea de Huntington. Revista Cubana de Genética Humana 2001;3: 1-15 [ Links ]
29. Murray J, Cuckle H, Taylor G, Hewison J. Screening for fragile X Syndrome: information needs for health planners. J Med Screen 1997;4: 60-94. [ Links ]
Correspondencia: MSc. Melissa Vásquez Cerdas, Código postal: 2060 San José. Correo electrónico: melissa.vasquez@ucr.ac.cr

