










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.53 n.2 San José Jun. 2011
Revisión
Linfohistiocitosis hemofagocítica, el espectro desde la enfermedad genética al síndrome de activación macrofágica
(Hemophagocityc Lymphohistiocytosis: A Spectrum from the Genetic Disorder to the Macrophage Activation Syndrome)
Servicio de Inmunología y Reumatología Pediátrica Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”
Abreviaturas: LHH, linfohistiocitosis hemofagocítica; LHHF, LHH familiar; cNK, células NK; TCD8, linfocitos T CD8+; SAM, síndrome de activación macrofágica; AIJs, artritis idiopática juvenil sistémica; CH, síndrome de Chediak-Higashi; SG, síndrome de Griscelli; XLP, síndrome linfoproliferativo ligado al cromosoma X; VEB, virus de Epstein-Barr; LHH-V, linfohistiocitosis hemofagocítica asociada a virus; LESj, lupus eritematoso sistémico juvenil.
Resumen
El compromiso de la regulación de la inflamación produce activación excesiva y expansión de macrófagos y linfocitos T que desencadenan una reacción inflamatoria severa, sin vías naturales de control. Los trastornos hemofagocíticos son la traducción clínica de este proceso inflamatorio. La linfohistiocitosis hemofagocítica se refiere a todas las variantes de esta patología, y el síndrome de activación macrofágica, a la variante asociada con enfermedad autoinmune. Los casos primarios se asocian con la forma familiar autosómica recesiva y los secundarios con inmunodeficiencias primarias, infección, malignidad y enfermedades autoinmunes. El principal distintivo de este grupo de patologías es la proliferación agresiva de macrófagos e histiocitos que fagocitan otras células sanguíneas. La reducción en la actividad de las células NK produce un aumento en la activación y expansión de linfocitos T, los cuales producen grandes cantidades de citoquinas. Las citoquinas inducen activación de macrófagos y células dendríticas, infiltración tisular y producción de interleuquinas, lo que genera una reacción inflamatoria severa, responsable del daño tisular y de las manifestaciones clínicas. El curso clínico se caracteriza principalmente por fiebre prolongada, hepatoesplenomegalia y citopenias. Los estudios de laboratorio muestran aumento de ferritina, triglicéridos e hipofibrinogenemia. La hemofagocitosis en médula ósea está presente en más del 80% de los casos al diagnóstico. El tratamiento está dirigido contra el linfocito T y los histiocitos hiperactivados, combinando quimioterapia con inmunosupresores y, en algunos casos, trasplante de células madre hematopoyéticas. Este tratamiento ha producido un cambio en la sobrevida de los pacientes. El protocolo de tratamiento HLH-2004 es una guía que estandariza el tratamiento, combinando etopósido, dexametazona y ciclosporina A. En Costa Rica se han reportado 60 casos en población pediátrica, con una mortalidad promedio del 44%.
Descriptores: linfohistiocitosis hemofagocítica, síndrome hemofagocítico, síndrome de activación macrofágica, lupus eritematoso sistémico juvenil, artritis idiopática juvenil sistémica, virus de Epstein-Barr, ferritina, hemofagocitosis, trasplante de médula ósea.
Abstract
Hemophagocytic lymphohistiocytosis is characterized by a severe hyperinflammatory condition, with macrophages and T cells activation and expansion, without regulatory pathways for the termination of the inflammatory activity. Hemophagocytic syndromes are clinical translation of an overwhelmed inflammatory response. The term hemophagocytic lymphohistiocytosis applies to all the variants of the syndrome and macrophage activation syndrome refers to the variant associated with autoimmune diseases. Primary cases are related to a familiar autosomic recessive disease and the secondary ones to primary immunodeficiencies, infection, malignancy and autoimmune diseases. Physiopathology of the disease is related mainly to the aggressive macrophage and histiocyte proliferation and phagocytosis of blood cells. An impaired function of NK cells and cytotoxic T-cells, increase cell activation and expansion and cytokine production inducing macrophage activation, tissue infiltration and tissue damage. The main symptoms are prolonged fever, cytopenias, hepatosplenomegaly, and hemophagocytosis. Biochemical markers include elevated ferritin and triglycerides and low fibrinogen. Bone marrow hemophagocytosis is present in more than 80% of the cases at diagnosis. Treatment targets activated T-cells an histiocytes, combining chemotherapy, immunosuppressors and in selected cases hematopoyetic stem cell transplantation. Treatment produced a positive change in patient survival. HLH-2004 treatment protocol is an standarized guideline that combine etoposide, dexamethasone and cyclosporine A. In Costa Rica 60 cases have been reported in children, with a 44% mortality.
Keywords: hemophagocytic lymphohistiocytosis, hemophagocytic syndrome, macrophage activation syndrome, juvenile-onset systemic lupus erythematosus, systemic juvenile idiopathic arthritis, Epstein-Barr virus, ferritin, hemophagocytosis, bone marrow transplantation.
La respuesta inflamatoria tiene mecanismos bien definidos para delimitar su duración y restablecer el equilibrio de la relación huésped-parásito, después de que el sistema inmune ha inducido inflamación. El compromiso de la regulación de la inflamación produce activación excesiva y expansión de macrófagos y linfocitos T que desencadenan una reacción inflamatoria severa, sin vías naturales de control. Los trastornos hemofagocíticos son la traducción clínica de este proceso inflamatorio y reflejan defectos que alteran la comunicación normal entre las respuestas del sistema inmune innato y adaptativo.1
En general, se ha relacionado esta patología con la edad pediátrica, probablemente por las descripciones iniciales de la forma familiar. Sin embargo, es un proceso inflamatorio que se puede presentar a cualquier edad, desde el recién nacido hasta el adulto mayor. Hay descripciones recientes del diagnóstico en adolescentes y adultos, de la forma familiar, en vinculación con cierto tipo de mutaciones.
El término correcto para llamar esta patología se ha prestado a confusión, en este artículo se usa linfohistiocitosis hemofagocítica, para referirse de manera inclusiva a todas las variantes del síndrome, y se deja síndrome de activación macrofágica, para indicar la variante asociada con enfermedades autoinmunes.
Clasificación
El término linfohistiocitosis hemofagocítica (LHH) fue acuñado por la “International Histiocyte Society” en 1998, para describir la LHH familiar (LHHF), una enfermedad genética con inflamación sistémica severa.2 Scott y Robb-Smith describieron el síndrome por primera vez en 1939, como reticulosis histiocítica medular.3 La descripción de LHHF es de Farquhar y Claireux, en 1952, quienes describieron la presentación pediátrica del cuadro con fiebre, hepatoesplenomegalia y citopenias.4 En el grupo de pacientes con enfermedades genéticas se incluyen los que tienen síndromes con inmunodeficiencia primaria.5
Los casos no familiares o adquiridos de LHH se describieron como secundarios y asociados con infecciones, malignidad, inmunosupresión y enfermedades autoinmunes. La estructura de clasificación, basada en las recomendaciones de la “Histiocyte Society” para las patologías del histiocito, se muestra en el Cuadro 1.6 En ocasiones, en publicaciones de casos en adultos se utiliza el término “síndrome de activación macrofágico reactivo”, para indicar LHH adquirida o síndrome hemofagocítico, sin tomar en cuenta la patología de fondo.7

El principal distintivo de este grupo de patologías es la proliferación agresiva de macrófagos e histiocitos que fagocitan otras células sanguíneas. Es un crecimiento de células descontrolado, sin características de malignidad y sin clonalidad, con migración ectópica, principalmente a bazo, ganglios linfáticos, médula ósea, hígado, piel y las membranas que cubren la médula espinal.
La teoría actual que explica la fisiopatología de esta enfermedad, identifica una reacción inmunológica anormal, como consecuencia de la activación de linfocitos T y de macrófagos, sin la intervención de los mecanismos de apoptosis normales, que conduce a inflamación sistémica. Es un problema de función más que de ausencia de estas células; la infiltración tisular puede producir una linfopenia TCD8+ paradójica. Una respuesta Th1 descontrolada y una función citotóxica defectuosa son los mecanismos fundamentales en la fisiopatología de este grupo de patologías.8-10
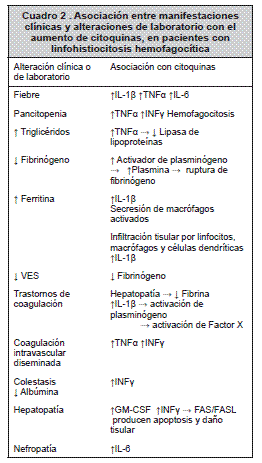
Se ha identificado una deficiencia en la función citotóxica de células NK (cNK) y linfocitos T CD8+ (TCD8). En el mecanismo normal de citotoxicidad, las cNK y los TCD8 destruyen sus células blanco utilizando una vía no secretora (FasL, CD95L) y una secretora, liberando gránulos citotóxicos que contienen perforinas, granzima y otras sustancias proteolíticas. La célula citotóxica y la célula blanco forman una sinapsis inmunológica en el punto de contacto, hacia el cual rota el centro de organización de microtúbulos, se liberan los gránulos citotóxicos que se fusionan con la membrana y liberan su contenido en la sinapsis, se producen poros en las membranas celulares y, como consecuencia, lisis osmótica, degradación proteica y apoptosis. 5, 11 La reducción en la actividad de cNK produce un aumento en la activación y expansión de linfocitos T, los cuales generan grandes cantidades de citoquinas (interferón gamma, factor de necrosis tumoral alfa, factor estimulante de colonias de granulocitos-macrófagos). Las citoquinas inducen activación de macrófagos y células dendríticas, infiltración tisular y producción de interleuquinas (IL-6, IL-2, IL-1β, IL-8, IL-10, IL-12, IL-18), lo que produce una reacción inflamatoria severa, responsable del daño tisular y de las manifestaciones clínicas. Cuando se determinan niveles de citoquinas en suero se encuentran concentraciones de 10 a 1000 veces mayores que las normales. 5, 10, 12-14
Los gatillos que disparan esta serie de eventos probablemente utilizan vías propias de cada uno de ellos (virus, bacterias, hongos, autoinmunidad, medicamentos). En los pacientes, durante la etapa activa de la enfermedad, se detectanniveleselevadosdevariascitoquinasproinflamatorias, las cuales producen alteraciones progresivas de diferentes órganos, que causan a la muerte del paciente. 15
Además, se puede presentar pérdida rápida de peso, sangrado por alteración de los tiempos de coagulación (prolongación de TP y TPT), infiltrados pulmonares y compromiso cardiaco y renal.5, 9, 10
La hematología muestra anemia arregenerativa y trombocitopenia de aparición temprana; la mitad de los casos tienen neutropenia, y la leucopenia es menos frecuente y más tardía en el curso de la enfermedad. Tres de cada cuatro pacientes sufren pancitopenia y todos, bicitopenia. En otros estudios de laboratorio se identifica un aumento de ferritina, triglicéridos, transaminasas, bilirrubinas y deshidrogenasa láctica e hipofibrinogenemia, que provoca disminución en la VES. El trastorno de la coagulación más frecuenteesladeficienciaaisladadefibrina,comoconsecuencia de alteraciones en la función hepática y por la activación por IL-1β de plasminógeno y de factor X. Aunque no es frecuente, pero sí asociada con alta mortalidad, se puede presentar coagulación intravascular diseminada por la sobreproducción de interferón gamma y de factor de necrosis tumoral alfa.10, 15
Citolisis y colestasis están asociadas con alteración de la función hepática. Interferón gama favorece el desarrollo de colestasis e hipoalbuminemia. La interacción Fas/Fas ligada en respuesta a la sobreproducción de interferón gama, explica la apoptosis y el daño del tejido hepático.
Se pueden detectar otras alteraciones asociadas con el proceso inflamatorio, como hipo o hipergammaglobulinemia, Coombs directo positivo e hiponatremia asociada a secreción inapropiada de hormona antidiurética.1
Algunas de las manifestaciones clínicas se pueden interpretar como la respuesta inflamatoria normal hacia un agente infeccioso, pero la severidad indica progresión y falta de respuesta al tratamiento debe hacer sospechar LHH.
El estudio del aspirado de médula ósea muestra evidencia de eritropoyesis inefectiva, con incremento de la eritropoyesis, pero elevación moderada de reticulocitos; la actividad prolongada del proceso inflamatorio puede producir una médula ósea aplásica. La hemofagocitosis en médula ósea está presente en más del 80% de los casos al diagnóstico, pero puede estar ausente al inicio del cuadro inflamatorio.10
La mortalidad de LHH se reporta entre un 22 y un 59%. Las variantes asociadas con malignidad o infección por VEB son las que tienen las tasas de mortalidad más altas. En los casos que fallecen, la muerte ocurre durante las primeras 4 a 8 semanas y está asociada con falla orgánica multisistémica, sangrado o sepsis.10
Criterios de diagnóstico
Como marcadores de diagnóstico se han identificado la elevación de la ferritina, de la cadena alfa del receptor soluble de IL-2 (sCD25), la hipofibrinogenemia y la deficiencia en la actividad de cNK. En estudios en población pediátrica, un nivel de ferritina mayor a 10 000 µg/L mostró una sensibilidad del 90% y una especificidad del 96% para LHH. 5, 18 La tasa de reducción en el nivel de ferritina se asocia con mortalidad; un paciente tiene 17 veces más probabilidad de fallecer cuando el descenso de ferritina es menor del 50%.19
Un grupo de hallazgos clínicos y de laboratorio fueron propuestos por la “Histiocyte Society” para facilitar el diagnóstico de LHH (Cuadro 3).20 Sin embargo, estos criterios pueden no tener la especificidad suficiente, en los casos cuando la enfermedad de fondo se acompaña de inflamación, como en los síndromes asociados a infección o a enfermedades autoinmunes. Para estos casos resulta más pertinente el uso de los criterios de diagnóstico propuestos para el síndrome de activación macrofágica (SAM) en artritis idiopática juvenil sistémica (AIJs) (Cuadro 4).21, 22


Para el diagnóstico clínico de LHH, encontrar hemofagocitosis en el aspirado de médula ósea no es un prerrequisito, a pesar de que es un marcador que indica histiocitos activados. Los estudios de autopsias han demostrado que la hemofagocitosis se encuentra con mayor frecuencia en hígado, bazo y ganglios linfáticos, que en médula ósea. Una población de células activadas se puede demostrar a pesar de la ausencia de hemofagocitosis.1
Síndromes con linfohistiocitosis hemofagocítica
LHH no es una enfermedad; se debe estudiar como un síndrome que se identifica asociado a varias enfermedades, las cuales conducen a un fenotipo semejante producido por inflamación severa.
Familiar: es la forma primaria, con etiología genética conocida, se hereda en forma autosómica recesiva y LHH es la única manifestación clínica. La incidencia se ha estimado en 0,12/100 000 niños por año y se reporta una tendencia a ser más frecuente en varones.9, 23 En reportes de cohortes, la mitad de los casos con un hermano afectado provienen de matrimonios consanguíneos. En un 70%-80% de los casos, las manifestaciones clínicas se inician antes de un año de edad; solamente el 10% de los casos es sintomático en las primeras 4 semanas de edad y muy pocos tienen síntomas al nacer. 9, 24 Se han reportado casos de inicio en la adolescencia y en adultos, en los cuales se identifica el daño molecular asociado con la enfermedad genética familiar.25, 26 Con la forma familiar se han asociado mutaciones en el gen de perforina (PRF1), y la deficiencia de perforina explica el 15%-20% de los casos de LHH. Otro gen estudiado es MUNC 13-4, esencial para la fusión de gránulos citolíticos con otras estructuras. Un mutación en el gen de sintaxina, que se asocia con defectos en la degranulación, explica otro grupo de casos familiares de LHH.1, 9
Asociado a inmunodeficiencias:las inmunodeficiencias que se han asociado con LHH son el síndrome de Chediak-Higashi (CH), el síndrome de Griscelli (SG) y el síndrome linfoproliferativo ligado al cromosoma X (XLP).1, 9, 27
En CH (AR, albinismo oculocutáneo, pelo plateado, inclusiones gigantes en los leucocitos) se describe como fase acelerada de la enfermedad a la presentación de LHH.28,29 En SG tipo 2 (AR, hipopigmentación, deficiencia funcional de neutrófilos), los episodios de LHH son frecuentes y asociados a la mortalidad de la enfermedad. 9,27,30 En XLP (XL, Purtilo) la infección por virus de Epstein-Barr (VEB) es el gatillo para LHH, que es la causa de muerte en la mitad de los casos.9, 27,31
Asociada a malignidad: en un estudio en adultos se describió la asociación con una incidencia anual de 0,36/100 000 individuos, un curso de LHH agresivo, con infección concomitante y mortalidad en la mitad de los casos.1, 9,32
LHH puede ocurrir antes o durante el tratamiento de la malignidad, o bien, ser la primera manifestación clínica del caso. Se considera como la asociación con el peor pronóstico; la peor sobrevida se ha reportado en linfomas T/NK. En los casos de linfoma no Hodgkin T con LHH, se ha descrito una sobrevida promedio de 40 días si no se da tratamiento específico para LHH. La asociación con malignidad es más frecuente en adultos que en niños. Hay reportes de casos con linfoma de Hodgkin y enfermedad de Castleman.1, 9, 10, 32
Asociada a infección: la primera descripción de esta asociación relacionó LHH con infección viral en adultos con trasplantes de órganos; los casos se identificaron como LHH asociado a virus (LHH-V), pero infecciones por bacterias, protozoarios y hongos también han sido asociados con LHH. En muchos de los casos no se identifica una inmunosupresión o inmunodeficiencia de fondo que explique la asociación entre infección y LHH.1, 10
Muchos virus pueden desencadenar LHH, sin embargo, más del 50% de los casos están asociados con virus herpes, y dentro de ellos, VEB es el más frecuente. LHH asociada con VEB es más común en población pediátrica y es de mal pronóstico, en especial en inmunocomprometidos. 33 Citomegalovirus contribuye con el 30%-50% de los casos de LHH-AV.Otrosvirusdescritossonherpessimplex, parvovirus, adenovirus, hepatitis, rubéola, respiratoriosincicial, coxsackie, influenza y VIH.1, 10 Dos casos fueron reportados durante la pandemia 2009 de influenza A H1N1.34
Histoplasmosis es la infección por hongos que se asocia con mayor frecuencia a LHH. Hay reportes frecuentes, también, de asociación con Leishmania. Se ha reportado la asociación con aspergillus, cándida y criptococo. Algunos casos se describieron en infecciones por Plasmodium, Pneumocystis jiroveci, toxoplasma, babesia, micobacterias, mycoplasma, Brucella, Borrellia, Ehrlichia y brucela 10, 35
Síndrome de activación macrofágica: se identifica como SAM a la LHH asociada con enfermedades autoinmunes; ha sido muy bien descrito en casos de AIJs y lupus eritematoso sistémico juvenil (LESj). El síndrome fue descrito en 1985 por Hadchouel en pacientes con AIJs, y el término SAM se propuso en 1993.1, 9, 10 12, 15
En un reporte de 26 casos con enfermedades sistémicas, se encontró asociado con lupus, artritis reumatoidea, síndrome de Sjögren, enfermedad de Kawasaki, poliarteritis nodosa, sarcoidosis, dermatomiositis, fiebres periódicas y enfermedad mixta de tejido conectivo. Sin embargo, muchos de estos casos tenían enfermedades infecciosas concomitantes. 10, 36
En un estudio multicéntrico en población pediátrica de 38 pacientes con LESj y SAM, se reportó la mayor sensibilidad y especificidad para hiperferritinemia, seguida por aumento de la deshidrogenasaláctica, hipertriglicerinemia e hipofibrinogenemia. El grupo de investigadores concluyó que la presencia de fiebre sin etiología y citopenia asociados con hiperferritinemia en un paciente con LESj, debe inducir la sospecha de SAM.37 En 9 casos pediátricos reportados en 2007, la edad tuvo un intervalo de 10 a 17 años; en la mitad
SAM asociado a AIJs es una causa importante de morbilidad y mortalidad. En dos series de casos se reportó una mortalidad del 8%-22%. En un estudio de 74 pacientes con AIJs con SAM, se señaló como marcadores a hemorragias, alteración de sistema nervioso central, plaquetopenia, aumento de aspartato aminotransferasa, leucopenia e hipofibrinogenemia.21 En 4 de 6 casos identificados en el norte de India, SAM fue la manifestación inicial de AIJs. 39
SAM es una complicación de difícil diagnóstico en AIJs, porque puede confundirse con una reactivación, con infecciones o con efectos adversos a los medicamentos. En el Cuadro 4 se muestra un grupo de parámetros para el diagnóstico de SAM en AIJs.7, 12, 21, 22
Tratamiento
La terapia para LHH actual está dirigida contra el linfocito T y los histiocitos hiperactivados, combinando quimioterapia proapoptótica con inmunosupresores. 9 Este tratamiento ha producido un cambio en la sobrevida de los pacientes, aumentándola del 5% al 50%-70%. Debido a que LHH puede evolucionar rápidamente y ser fatal, el tratamiento específico debe iniciarse cuando hay suficiente sospecha clínica. 1 La mejoría del cuadro inflamatorio se puede monitorear con los niveles de ferritina.17, 40
El uso del protocolo HLH-94 de la “Histiocyte Society” se ha generalizado, a pesar de que fue diseñado con base en la LHHF.41 Estas guías de tratamiento tenían como propósito obtener remisión del estado de inflamación y curar en forma definitiva con trasplante alogénico de células madre hematopoyéticas. HLH-94 combina etoposido iv / dexametazona iv o vo y terapia de mantenimiento con ciclosporina A.1, 9, 20 En los pacientes con LHH que recibieron HLH-94, un 75% obtuvieron remisión clínica después de 8 semanas de tratamiento Una cuarta parte de los pacientes tuvieron síntomas persistentes y fallecieron por complicaciones del LHH. La sobrevida libre de LHH a 3 años, de los pacientes tratados, fue del 55%.1 La terapia intratecal se recomienda en pacientes con signos persistentes de actividad en sistema nervioso central o en su reactivación.20
El uso de HLH-94 como tratamiento de LHH se ha reportado efectivo en pacientes con XLP, CH, SG, LHH asociado a VEB y SAM.7-9, 15, 33, 42, 43
Una modificación de HLH-94, HLH 2004 se recomienda como el protocolo de tratamiento para los diferentes tipos de LHH.1, 20, 33
Los pacientes que después de controlar la reacción inflamatoria con inmunosupresores y quimioterapia (HLH94), reciben un trasplante alogénico de células madre hematopoyéticas con donador idéntico, tienen una sobrevida libre de enfermedad a 3 años, del 70%.1, 43-47
Otras terapias como inmunoglobulina iv a altas dosis, ciclosporina A, globulina anti-timocítica, biológicos anti-TNF, Campath-1H, abatacept, alemtuzumab y anakinra que suprimen macrófagos y linfocitos T activados, se han reportado efectivas en series pequeñas de casos. 1, 7, 9,48, 49
Al escoger el tipo de tratamiento se debe tomar en cuenta la posibilidad de que se trate de un caso de LHHF. Niños menores de 1 año de edad, con posibilidades de tener enfermedad genética y todos los casos con signos y síntomas severos, son candidatos para terapia combinada con dexamentasona, ciclosporina A y etopósido (HLH-94, HLH2004). Los casos menos graves pueden responder a esteroides e inmunoglobulina iv. Pero principalmente en niños se debe tener presente que si la evolución lo amerita, se debe utilizar etopósido. El riesgo a los efectos adversos de etopósido es preferible al riesgo de perder un paciente por un tratamiento inadecuado o insuficiente. En casos de SAM, esteroides con o sin ciclosporina A, se han reportado como suficientes para controlar el cuadro inflamatorio. 1, 9, 15, 20
LHH en Costa Rica
En población pediátrica costarricense se han realizado dos estudios retrospectivos, uno en 2000, de Castillo-Salas y Porras, de 19 casos, la mayoría asociados a VEB, en el que se reportó una edad promedio al diagnóstico de 3, 7 años (0,7-11,6 años) y una mortalidad del 46, 4%. 50
El otro en 2011, de Lavagni y Porras (comunicación personal), de 41 casos, la mayoría asociados a infección (78%), el 41,5% menores de 2 años y con mortalidad del 48,8%.
Referencias
1. Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin N Am 2008; 28: 293-313. [ Links ]
2. Henter JI, Arico M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998; 12: 417-433. [ Links ]
3. Scott R, Robb-Smith A. Histiocytic medullary reticulosis. Lancet 1939; 2: 194-198. [ Links ]
4. Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child 1952; 27: 519-525. [ Links ]
5. Janka G. Hemophagocytic lymphohistiocytosis: when the immune system runs amok. Klin Padiatr 2009; 221: 278-285. [ Links ]
6. Arceci RJ. The histiocytoses: the fall of the tower of Babel. Eur J Cancer 1999; 35: 747-769. [ Links ]
7. Tristano AG. Macrophage activation syndrome: a frequent but under-diagnosed complication associated with rheumatica diseases. Med Sci Monit 2008; 14: 27-36. [ Links ]
8. Bhattacharyya M, Ghosh MK. Hemophagocytic lymphohistiocytosis – Recent concept. J Assoc Physicians India 2008; 56: 453-457. [ Links ]
9. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr 2007; 166: 95-109. [ Links ]
10. Créput C, Galicier L, Buyse S, Azoulay E. Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intensive Care Med 2008; 34: 1177-1187. [ Links ]
11. Stinchcombe J, Bossi G, Griffiths G. Linking albinism and immunity: the secrets of secretory lysosomes. Science 2004; 305: 55-59. [ Links ]
12. Grom AA, Mellins ED. Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol 2010; 22: 561-566. [ Links ]
13. Menasche G, Feldmann J, Fischer A, Basile Gde S. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev 2005; 203: 165-179. [ Links ]
14. Egeler RM, Shapiro R, Loechelt B, Filipovich A. Characteristic immune abnormalities in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 1996; 18: 340-345. [ Links ]
15. Janka GE. Hemophagocytic syndromes. Blood Rev 2007; 21: 245-253. [ Links ]
16. Henter JI, Elinder G. Cerebromeningeal haemophagocytic lymphohistiocytosis. Lancet 1992; 339: 104-109. [ Links ]
17. Howells DW, Strobel S, Smith I, Levinsky RJ, Hyland K. Central nervous system involvement in the erythrophagocytic disorders of infancy: the role of cerebroespinal fluid neopterins in their differential diagnosis and clinical management. Pediatr Res 1990; 28: 116-119. [ Links ]
18. Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2008; 50: 1227-1235. [ Links ] 19. Lin T, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer 2011; 56: 154-155 [ Links ] 20. Henter JI, Horne AC, Aricó M, Egeler RM, Filipovich A, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. [ Links ] 21. Ravelli A, Magni-Manzoni S, Pistorio A, Besana C, Foti T, Ruperto N, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr 2005; 146: 598-604. [ Links ] 22. Davì S, Consolaro A, Guseinova D, Pistorio A, Ruperto N, Martini A, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol 2011; 38: 1-5. [ Links ] 23. Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paeditr Scand 1991; 80: 428-435. [ Links ] 24. Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia 1996; 10: 197-203. [ Links ] 25. Allen M, De Fusco C, Legrand F, Clementi R, Conter V, Danesino C, et al. Familial hemophagocytic lymphohistiocytosis: how late can onset be? Haematologica 2001; 86: 499-503. [ Links ] 26. Clementi R, Emmi L, Maccario R, Liotta F, Moretta L, Danesino C, et al. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood 2002; 100: 2266-2267. [ Links ] 27. Trottestam H, Beutel K, Meeths M, Carlsen N, Heilmann C, Pašić S, et al. Treatment of the X-linked lymphoproliferative, Griscelli and Chédiak-Higashi syndromes by HLH directed therapy. Pediatr Blood Cancer 2009; 52: 268-272. [ Links ] 28. Bejaoui M, Veber F, Girault D, Gaud C, Blanche S, Griscelli C, et al. The accelerated phase of Chediak-Higashi syndrome. Arch Fr Pediatr 1989; 46: 733-736. [ Links ] 29. Ortuño FJ, Fuster JL, Jerez A. Síndrome de Chediak-Higashi. Med Clin (Barc) 2010; 135: 512-518. [ Links ] 30. Van Gele M, Dynoodt P, Lambert J. Griscelli syndrome: a model system to study vesicular trafficking. Pigment Cell Melanoma Res 2009; 22: 268-282. [ Links ] 31. Rezaei N, Mahmoudi E, Aghamohammadi A, Das R, Nichols KE. X-linked lymphoproliferative syndrome: a genetic condition typified by the triad of infection, immunodeficiency and lymphoma. Br J Haematol 2011; 152: 13-30. [ Links ] 32. Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma 2011: en prensa. [ Links ] 33. Imashuku S. Treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis (EBV-HLH); update 2010. J Pediatr Hematol Oncol 2011; 33: 35-39. [ Links ] 34. Őzdemir H, Çiftçi E, Ünal İnce E, Ertem M, İnce E, Doğru Ü. Hemophagocytic lymphohistiocytosis associated with 2009 pandemic Influenza A (H1N1) virus infection. J Pediatr Hematol Oncol 2011; (en prensa). [ Links ] 35. Hanson D, Walter AW, Powell J. Ehrlichia-induced hemophagocytic lymphohistiocytosis in two children. Pediatr Blood Cancer 2011; [ Links ]
36. Dhote R, Simon J, Papo T, Detournay B, Sailler L, Andre MH, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum 2003; 49: 633-639. [ Links ]
37. Parodi A, Davì S, Pringe A, Pistoro A, Ruperto N, Magni-Manzoni S, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus. A multinational multicenter study of thirty-eight patients. Arthritis Rheum 2009; 60: 3388-3399. [ Links ]
38. Pringe A, Trail L, Ruperto N, Buoncompagni A, Loy A, Breda L, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: an under-recognized condition? Lupus 2007; 16: 587-592. [ Links ]
39. Singh S, Chandakasan S, Ahluwalia J, Suri D, Rawat A, Ahmed N, et al. Macrophage activation syndrome in children with systemic onset juvenile idiopathic arthritis: clinical experience from northwest India. Rheumatol Int 2011; (en prensa) [ Links ]
40. Knovich MA, Storey JA, Coffman LG, Torti SV, Torti FM. Ferritin for the clinician. Blood Rev 2009; 23: 95-104. [ Links ]
41. Henter JI, Aricò M, Egeler RM, Elinder G, Favara BE, Filipovich AH, et al. HLH-94: a treatment protocol for hemophatocytic lymphohistiocytosis. Med Pediatr Oncol 1997; 28: 342-347 [ Links ]
42. Lee JS, Kang JH, Lee GK, Park HJ. Successful treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis with HLH-94 protocol. J Korean Med Sci 2005; 20: 209-214. [ Links ]
43. Ishii E, Ohga S, Imashuku S, Kimura N, Ueda I, Morimoto A, et al. Review of hemophagocytic lymphohistiocytosis (HLH) in children with focus on japanese experiences. Crit Rev Oncol Hematol 2005; 53: 209-223. [ Links ]
44. Horne A, Janka G, Maarten ER, Gadner H, Imashuku S, Ladisch S, et al. Haematopoietic stem cell transplantation in haemophagocytic limphohistiocytosis. Br J Haematol 2005; 129: 622-630 [ Links ]
45. Ouachee-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, et al. Hematopietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics 2006; 117: 743-750. [ Links ]
46. Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood 2006; 107: 1233-1236. [ Links ]
47. Yoon HS, Im HJ, Moon HN, Lee JH, Kim HJ, Yoo KH, et al. The outcome of hematopoietic stem cell transplantation in Korean children with hemophagocytic lymphohistiocytosis. Pediatr Transplant 2010; 14: 735-740. [ Links ]
48. Strout MP, Seropian S, Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic limphohistiocytosis. Nat Rev Clin Oncol 2010; 7: 415-420. [ Links ]
49. Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol 2011; 17: 23-27. [ Links ]
50. Castillo-Salas S. Síndrome de activación macrofágica en niños. Tesis para licenciatura. UNIBE: San José, pp: 1-97, 2000. [ Links ]

