










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.53 n.1 San José Jan. 2011
Caso clínico
1Servicios de Inmunología y Reumatología
2Servicio de Citogenética3Servicio de Cardiología Hospital Nacional de Niños “Dr.Carlos Sáenz Herrera” Caja Costarricense de Seguro Social.
Abreviaturas: SD22q11, síndrome con deleción 22q11; VCFS, síndrome velocardiofacial; FISH, hibridización in situ con fluorescencia; SDG, secuencia secundaria de Di George.
Correspondencia: Dr. Oscar Porras, Inmunología y Reumatología, Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera”, Apartado 1654, 1000 San José, Costa Rica.
Correo electrónico: porrasza@racsa.co.cr
Resumen
El síndrome con deleción 22q11 es una enfermedad autosómica recesiva causada por una microdeleción 22q11.2. En este artículo se reportan los tres primeros casos del síndrome confirmados por citogenética en Costa Rica. El estudio de fluorescencia con hibridización in situ que demostró la microdeleción 22q11.2, se indicó por la sospecha clínica del síndrome, en 2 niños y una niña con malformaciones congénitas conotruncales de corazón. Dos de los casos se encuentran vivos a la fecha cuando se escribió este reporte y uno falleció en el postoperatorio inmediato de la cirugía para corregir la cardiopatía. Al inicio de los síntomas, en los tres casos se documentó falla para progresar y en dos se anotó dismorfismo en referencia a rasgos faciales anormales. En un caso se reportó paladar hendido y en otro pie, bott. A pesar de que la malformación congénita de corazón es el hallazgo clínico que con frecuencia induce al médico a pensar en este síndrome, los trastornos cognitivos y del comportamiento son las manifestaciones fenotípicas más frecuentes.
Descriptores: síndrome con deleción 22q11, síndrome velocardiofacial, secuencia de Di George, cardiopatía congénita, paladar hendido, inmunodeficiencia, FISH
Abstract
Key words: 22q11 deletion syndrome, Velo-cardio-facial syndrome, Di George sequence, congenital heart disease, cleft palate, immunodeficiency, FISH.
El Síndrome con Deleción 22q11 (SD22q11) es una enfermedad hereditaria con un patrón autosómico recesivo. La causa genética específica se encontró en 1992, al describirse una microdeleción en la banda q11.2 del cromosoma 22. Se describió como síndrome velocardiofacial (VCFS) en 1978, en 12 pacientes. Sin embargo, hay reportes de casos desde 1955.1 En 1968, Di George describió la asociación con aplasia de timo, hipoparatiroidismo y cardiopatía congénita.2,3 Recientemente se ha reportado la asociación con problemas cognoscitivos y del comportamiento.1,2,4 Los datos de incidencia y prevalencia son variables y asociados a la experiencia con SD22q11 de los médicos. Oskardottir y cols5 reportaron, en una región de Suecia, incidencia de entre 18,1 y 23,4 por 100.000 nacidos vivos, y prevalencia de entre 13,2 y 23,3 por 100.000 niños menores de 16 años. En ambos casos, los valores más altos se asociaron a la zona de trabajo del equipo con mayor experiencia en la enfermedad. Un tercio de los casos no tiene cardiopatía congénita y la mayoría no tiene paladar hendido.6,7
El diagnóstico molecular se logra por fluorescencia con hibridización in situ (FISH) con sondas moleculares específicas para la región del cromosoma 22 con la deleción. No se ha demostrado correlación entre la extensión de la deleción y la severidad de las manifestaciones clínicas. En la actualidad se considera que un individuo con deleción en la banda q11.2 en el cromosoma 22, tiene la enfermedad.7,8 El fenotipo muestra una alta variabilidad; como lo indica Shprintzen7: “ninguna de las características clínicas del síndrome ocurre en el 100% de los casos y no hay ningún caso reportado que tenga todas o la mayoría de los hallazgos clínicos. El diagnóstico por lo tanto está definido por la presencia de la deleción de ADN en la banda q11.2 del cromosoma 22”.7
Caso clínico
En este reporte se describen tres casos de pacientes con manifestaciones fenotípicas de SD22q11 (Cuadro 1), en los cuales la introducción, en 2006, de la tecnología FISH en el Laboratorio de Citogenética del Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera” (HNN), permitió el diagnóstico.
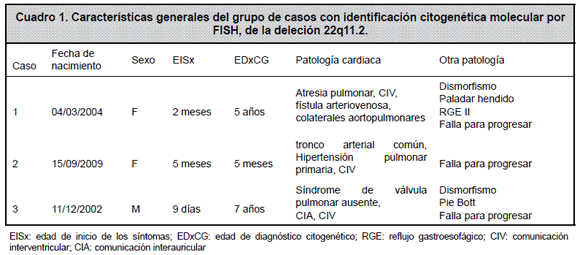
Caso 1.
Una niña de 2 meses de edad admitida en el HNN en mayo de 2004, referida de la Clínica Marcial Fallas Díaz, por un cuadro de tos, dificultad respiratoria, cianosis peribucal -principalmente al llorar-, y por la auscultación de un soplo sistólico III/VI. Al ingreso se identificaron los siguientes problemas: falla para progresar, cardiopatía congénita cianógena, dismorfismo y paladar hendido (palatoplastia a los 2 años y 5 meses). La radiografía de tórax mostró infiltrados paracardiacos y cardiomegalia. Un esofagograma evidenció un mecanismo de deglución normal con un reflujo gastroesofágico grado II.
Por la presencia de una cardiopatía congénita cianógena con dismorfismo y paladar hendido, se planteó como diagnóstico un VCFS. La evaluación cardiológica mostró atresia pulmonar con comunicación interventricular grande de mal alineamiento, y colaterales mayores aortopulmonares. Se le realizaron intervenciones quirúrgicas por su cardiopatía, con resultado satisfactorio a los 3 meses y a los 2 años de edad.
En abril de 2006, fue referida para evaluación inmunológica, por una historia de infección recurrente de vías respiratorias, bronquiolitis y hospitalizaciones por bronconeumonía en 4 ocasiones. El registro de infecciones no mostró compromiso en piel, tracto gastrointestinal, tracto urinario, ni episodios de septicemia o meningitis. Las bronconeumonías se asociaron con la identificación de virus Parainfluenza 3 y virus Respiratorio Sincicial. Los estudios inmunológicos mostraron: inmunoglobulinas séricas y subclases de inmunoglobulina G normales para la edad, presencia en suero de anticuerpos específicos contra toxoides tetánico y diftérico y contra antígenos de varicela y un ELISA-VIH no reactivo. El hemograma mostró hemoglobina y conteos de plaquetas y leucocitos normales. La cuantificación de poblaciones de linfocitos en sangre periférica realizada a los 2, 3 y 5 años, mostró inversión de la tasa CD4/CD8, con conteos normales de linfocitos T totales, T CD4+, TCD8+ y B.
A la edad actual de 6 años, es una niña eutrófica sin evidencia de hipocalcemia, que asiste a la escuela, recibe terapia de lenguaje y está en control por cardiopatía congénita, infección recurrente de vías respiratorias y escoliosis toracolumbar. A los 5 años de edad se le realizó el estudio citogenético de FISH que identificó la deleción 22q11.2.
Caso 2.
Una niña de 5 meses, referida al HNN por un cuadro de 3 días de evolución con disnea, tos productiva, cianosis y dificultad respiratoria. En el hospital de referencia se realizó una radiografía de tórax que mostró un infiltrado paracardiaco derecho y cardiomegalia. A su ingreso se encontró una paciente de 4,9 Kg, con frecuencia cardiaca de 167 latidos por minuto y frecuencia respiratoria de 60 respiraciones por minuto, afebril, con saturación del 88% en ambiente de oxígeno al 60%. Se auscultó un soplo plurifocal grado III/VI con galope, que se irradiaba a las carótidas. No se describió hepatoesplenomegalia. Con el ecocardiograma se estableció el diagnóstico de tronco arterial común tipo 1. Los estudios complementarios mostraron un hemograma con hemoglobina y conteos de plaquetas y leucocitos normales, calcio 8,9 mg/ dL (normal 9-11 mg/dL), calcio iónico 3,10 mg/dL (normal 4-5,2 mg/dL). La paciente fue manejada en la Unidad de Cuidados Intensivos; 5 días después de su ingreso se le realizó corrección quirúrgica de su cardiopatía congénita cianógena y falleció en el postoperatorio inmediato. El tiempo de estancia de esta paciente no permitió que se efectuaran estudios de la función inmune, pero sí el estudio citogenético de FISH a los 5 meses de edad, que mostró la deleción 22q11.2.
Caso 3.
Un niño a la edad de 15 días, referido al Servicio de Genética por dismorfismo, cardiopatía congénita cianógena y pie bott derecho. El genetista describió un niño con occipucio prominente, cuello corto y plicatus del pulgar, con un cariotipo normal. La evaluación cardiológica mostró cianosis, soplo sistólico y diastólico grado III/VI, pulsos normales. El ecocardiograma mostró un síndrome de válvula pulmonar ausente, con comunicación interventricular grande, de mal alineamiento de 12 mm, comunicación interauricular tipo ostium secundum de 12 mm con corto circuito de derecha a izquierda. El gradiente sistólico pico a través del tracto de salida del ventrículo derecho era de 100 mmHg. La arteria pulmonar principal y la arteria pulmonar derecha estaban dilatadas, con un diámetro de 22 mm cada una. La arteria pulmonar izquierda se encontró hipoplásica y medía apenas 5 mm de diámetro. A la edad de 7 años y 6 meses regresó a la consulta con historia de haber cursado sin infección recurrente, ni limitaciones de su actividad física, con saturación de oxígeno del 88% en aire ambiente y cianosis leve. No tiene estudios inmunológicos, ni de calcemia. Entre los estudios complementarios enviados tiene un segundo cariotipo normal y un estudio citogenético FISH a los 7 años, que identificó la deleción 22q11.2.
Citogenética.
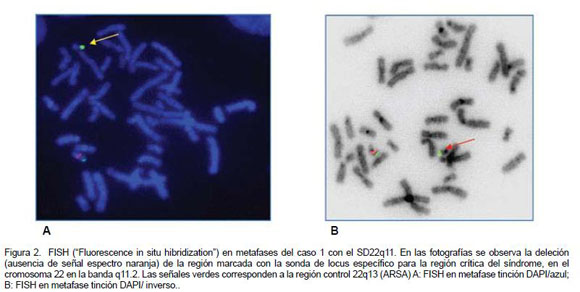
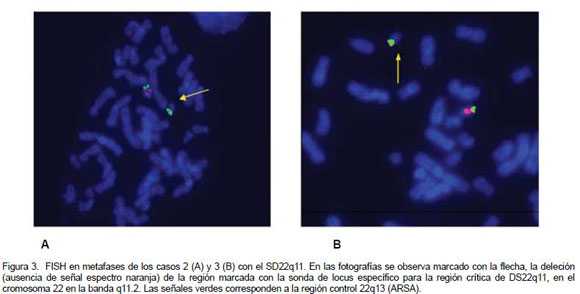
Estos tres pacientes se beneficiaron del hecho de que a partir de 2006 está disponible, en el Laboratorio de Citogenética del HNN, la prueba de FISH con la sonda Di George TUPLE 1, para identificar la microdeleción 22q11.2. El hallazgo de cardiopatía congénita compatible con VCFS motivó que se efectuara la prueba de FISH en los tres casos. Con tal propósito se realiza un cultivo de linfocitos de sangre periférica en medio de cultivo PB –MAX para cariotipo y FISH para la región 22q11.2.1 Para el FISH se utiliza la sonda de ADN, Vysis Di George Region Probe - LSI TUPLE 1 (espectro naranja) /LSI ARSA (espectro verde) en células en metafase e interfase obtenidas del cultivo de linfocitos en sangre periférica. El FISH se realiza por un proceso de desnaturalización de la hebra de ADN e hibridación por 20 horas, para cada sonda de ADN, según protocolo estandarizado en el Laboratorio de Citogenética.9
Se realiza un cariotipo sincronizado (cromosomas más largos) de sangre periférica con bandeo tripsina-Giemsa, con análisis de 20 mitosis (Figura 1). La evidencia de la microdeleción 22q11.2 se documenta con fotografía del cariotipo en un sistema computarizado Leyca (Figuras 2 y 3). Los cromosomas se clasifican de acuerdo con el sistema de nomenclatura ISCN 1995 (Figura 1). Para el FISH, la sonda LSI Vysis Di George Region Probe - LSI TUPLE 1 hibridiza en la banda 22q11.2 que fluoresce en espectro naranja y en la región LSI ARSA espectro verde de la banda 22q13. La sonda fluoresce de luz moderada a intensa, tanto en el núcleo de células en interfase, como en cromosomas en metafase. En células en interfase y metafase normales, las señales de las sondas verde y naranja generalmente aparecen en ambos cromosomas 22. En metafases anormales (deleción 22q11.2) aparece un cromosoma 22 con la señal naranja, y el otro sin la señal naranja, indicando la ausencia de la región cromosómica 22q11.2. En ambos cromosomas 22 se observa la señal verde como control de la región ARSA en 22q13. La evidencia de la microdeleción 22q11.2 se documenta con fotografía del FISH en un sistema computarizado Leyca (Figuras 2 y 3).
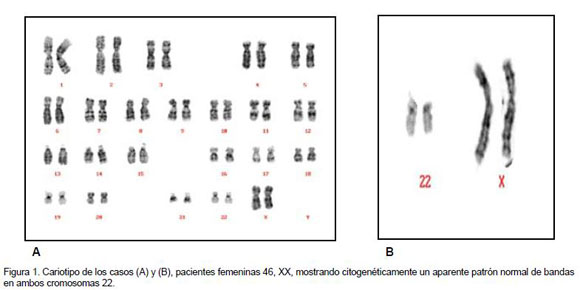
En el cariotipo se analizan 20 mitosis, según la clasificación ISCN 1995. El cariotipo en ambos casos mostró un patrón normal cromosómico femenino 46, XX en los casos 1 y 2, y masculino 46, XY en el caso 3 (Figura 1).
Los resultados de FISH mostraron en cada uno de los casos, de 15 a 20 metafases con un cromosoma 22 sin la señal para espectro rojo, lo cual demuestra la presencia de la microdeleción 22q11.2. La pérdida de genes ubicados en esta región 22q11.2 confiere las características fenotípicas en los tres pacientes descritos y permite el diagnóstico de SD22q11 (Figuras 2 y 3).
Discusión
La aplicación de la técnica de FISH para identificar la deleción 22q11.2 en pacientes con fenotipo de SD22q11, ha permitido concluir que los fenotipos reportados como: VCFS, secuencia de Di George, síndrome de Sedlačková, síndrome de anomalías conotruncales y de la cara, síndrome de Cayler y CATCH 22, son descripciones de la misma patología en una entidad clínica que expresa un fenotipo variable.1,3,10
El marcador molecular de este síndrome es la microdeleción hemizigota del segmento q11.2 en el brazo largo del cromosoma 22. Esta deleción es una de las anomalías cromosómicas más comunes, con una frecuencia de 1:2000 a 1:4000 en la población general. 4,11 Un 85-90% de los casos con SD22q11.2 tiene un deleción típica de ~3 millones de pares de bases, que incluyen ~40 genes, 10-12% tienen una deleción de ~1,5 millones de pares de bases y unos pocos muestran pequeñas deleciones. La mayoría de los casos son deleciones de novo, sin embargo, un 5-15% de los pacientes muestran una enfermedad autosómica dominante.8
La presencia de dismorfismo o cardiopatía congénita cianógena con una anomalía conotruncal en los tres casos reportados, indujo a efectuar el estudio citogenético que permitió demostrar la deleción diagnóstica en la banda q11.2 del cromosoma 22.
Los pacientes que tienen la secuencia secundaria de Di George (SDG) presentan también una facies característica, tetania por hipocalcemia, inmunodeficiencia congénita de la célula T e hipoplasia de timo. Además, la anatomía patológica muestra defectos en las glándulas paratiroides, en el arco aórtico y cardiaco. El espectro de la inmunodeficiencia varía entre una inmunodeficiencia severa y un sistema inmune normal.4 En dos de los casos reportados no se realizó un estudio inmunológico; en el otro se demostró una inversión de la tasa CD4/CD8 asociada a infección recurrente de vías respiratorias, pero no se encontró hipocalcemia, por lo que se debe concluir que entre los casos reportados solo se puede sospechar un caso de SD22q11 con SDG. El manejo de los casos se basa en el tratamiento médico quirúrgico de la cardiopatía y del paladar hendido, así como en la atención de los problemas de comportamiento, cognitivos y de aprendizaje.
La disponibilidad de la técnica de FISH para identificar la microdeleción 22q11.2, permite un diagnóstico citogenético molecular de los SD22q11, y la posibilidad de que reciban un manejo sindrómico más allá del tratamiento de la cardiopatía y el paladar hendido, lo cual posibilitará que estos niños disfruten de una mejor calidad de vida al recibir un abordaje interdisciplinario que incluya las áreas escolar y del comportamiento.
Referencias
1. Gothelf D, Frisch A, Michaelowsky E, Weizman A Shprintzen RJ. Velo-Cardio-Facial Syndrome. J Ment Health Res Intellect Disabil 2009; 2:149-167. [ Links ]
2. Di George AM, Lischner HW, Dacou C, Arey JB. Absence of the thymus. Lancet 1967; 1:1387. [ Links ]
3. Sullivan KE. Chromosome 22q11.2 deletion syndrome: Di George syndrome/velocardiofacial syndrome. Immunol Allergy Clin N Am 2008; 28: 353-366. [ Links ]
4. Sullivan KE. Di George Syndrome and Chromosome 22q11.2 deletion syndrome. En: Stiehm ER, Ochs HD, Winkelstein JA, eds. Immunologic disorders in infants and children. 5thedition. Philadelphia: Elsevier Saunders, 2004: 523-530. [ Links ]
5. Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child 2004; 89:148-151. [ Links ]
6. Oskarsdóttir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr 2005; 164: 146-153. [ Links ]
7. Shprintzen RJ. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev 2008; 14:3-10. [ Links ]
8. Emanuel BS. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev Disabil Res Rev 2008; 14: 11-18. [ Links ]
9. Manual de procedimientos del Laboratorio de Citogenética-Hospital Nacional de Niños. [ Links ]
10. Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndrome. Lancet 2007; 370:1443-1452. [ Links ]
11. The Velo-Cardio-Facial Syndrome Educational Foundation. Velocardio-facial syndrome: specialist fact sheet. En: http://www.vcfsef. org/pdf/VCFS_Factsheet_07.pdf. Consultado el 28 de octubre de 2010. [ Links ]
12. Butts SC. The facial phenotype of the velo-cardio-facial syndrome. Int J Pediatr Otorhinolaryngol 2009; 73: 343-350. [ Links ]
13. Oskarsdóttir S, Holmberg E, Fasth A, Strömland. Facial features in children with the 22q11 deletion syndrome. Acta Paediatr 2008; 97: 1113-1117. [ Links ]

