










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.51 n.2 San José Apr. 2009
Osteogénesis imperfecta con manifestaciones en el periodo neonatal
(Neonatal Presentation of Osteogenesis Imperfecta)
Gilberto Rodríguez-Herrera,1 María Jesús Navarro-Charpantier2
Resumen:
Se reporta un caso de un paciente masculino de un día de vida extrauterina; producto de una madre de 20 años, primigesta, prima segunda de su pareja. Nace por cesárea por presentación pélvica, con líquido amniótico meconizado, con un peso al nacer de 2275 gramos (RNTPEG). Al examen físico el niño se encontraba flácido, con cianosis leve, fontanelas amplias con comunicación de la anterior con la posterior, ausencia de escama occipital, escleras azules, retrognatia, extremidades cortas y con crepitación al movimiento. En las radiografías óseas con fracturas múltiples, formación de callo óseo y cambios displásicos en metáfisis. Se diagnostica por parte de los genetistas una osteogénesis imperfecta (OI) y se da consejo genético a los padres. La OI es un conjunto de trastornos genéticos que afectan la integridad del tejido conectivo, debido a que se presentan mutaciones en la síntesis del colágeno, ya sean autosómicas dominantes o recesivas. En vista de que el diagnóstico es predominante-mente clínico y radiológico, se debe profundizar en los patrones óseos, ya que los pacientes pueden desarrollar cambios quísticos, densos o frágiles. A partir de este caso de OI tipo 2 severa se pretende discutir las diferencias entre los diferentes grupos.
Descriptores: osteogénesis imperfecta, neonatos, protocolágeno tipo I.
Abstract:
We report the case of a male patient with one day of life, his mother is a 20-yr-old gravida 1 para 1 male, there is parental consanguinity (second cousin). This patient born alive by Caesarean section in breech presentation with meconium stained amniotic fluid and birthweight 2275 g (small for gestational age term infant). Physical examination findings in this case are hypotonia, mild cyanosis, large anterior fontanelle, absence of occipital scale, blue sclera, retrognathia and shortening of the long bones whit crepitation. The radiografphic findings show numerous fractures, hyperplastic callus formation and dysplastic changes in metaphyses. He was diagnosed by the geneticists with OI and given genetic counseling to parents. The osteogenesis imperfecta has a genetic background that affects connective tissue integrity, associated with collagen synthesis mutations, being dominant or recessive autosomic inheritance. In the majority of cases, diagnosis of IO is easy on the basis of clinical and radiological findings; for this reason must be important to emphasize on bone structure studies, because patients may develop cystic, dense or fragile changes. This case presents a patient who has osteogenesis imperfecta type II; we pretend to discuss the differences between Ols types.
Key words: osteogenesis imperfecta, neonates, type I protocollagen.
Caso clínico
Se trata de un paciente masculino de un día de vida extrauterina, vecino de Corralillo de Cartago; producto de una madre de 20 años, primigesta (G1P1C1AO), quien acudió al control prenatal en ausencia de patologías durante el embarazo. El parto fue quirúrgico por medio de cesárea el 16 de julio de 2007, en el Hospital Max Peralta de Cartago; el niño se encontraba en presentación pélvica a las 38 semanas de edad gestacional; se describe que el líquido amniótico fue meconizado. El niño tuvo un peso al nacer de 2275 gramos, con una talla de 40 cm (pequeño para edad gestacional en peso y talla) y circunferencia cefálica de 33 cm; además, se catalogó con una puntuación de APGAR de seis al primer minuto, ocho a los cinco minutos y diez a los diez minutos. Se describe que al nacer el niño se encontraba flácido con cianosis leve y extremidades cortas. Le envían una radiografía de tórax que evidencia el parénquima pulmonar sin alteraciones, y fracturas en la clavícula izquierda, séptimo arco costal y húmero izquierdo; por estos hallazgos es trasladado al Hospital Nacional de Niños Dr. Carlos Sáenz Herrera con diagnóstico presuntivo de acondroplasia. Los hallazgos del examen físico al ingreso mostraron un paciente alerta, consciente, activo, eupneico, hidratado, con presión arterial de 71/52 mmHg, frecuencia cardíaca de 166 latidos por minuto, frecuencia respiratoria de 42 respiraciones por minuto y temperatura de 36.6°C. Se describen las fontanelas amplias con comunicación de la anterior con la posterior, ausencia de escama occipital, escleras azules, pupilas isocóricas normoreactivas, con movimientos extraoculares adecuados y retrognatia. La exploración cardiopulmorar no sugería alteraciones llamativas; con adecuada expansión torácica, únicamente presentaba dolor a la palpación de la parrilla costal bilateral. El abdomen tenía peristalsis, se notaba globoso, timpánico, sin visceromegalias, pero con separación de músculos rectos abdominales. Tenía genitales masculinos normales con testículos en escroto y ano permeable. En ese momento llamaba mucho la atención que ambas extremidades superiores eran cortas, con crepitación ósea, con leve deformidad en el miembro superior izquierdo, descrita como dorsiflexión de la muñeca con limitación de la extensión, inadecuada pronosupinación del antebrazo y, en general, poca movilidad de la mano izquierda (Figura 1). Las extremidades inferiores se describen cortas con leve deformidad en genuvaro; presentan edema, adecuada movilidad espontánea y tono, predominantemente en flexión.
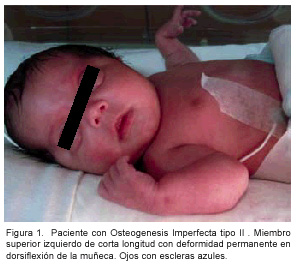
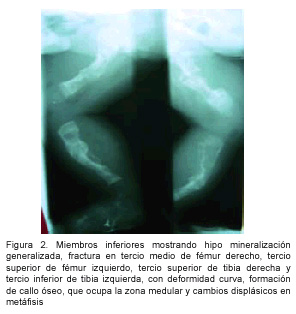
Los resultados de las pruebas de laboratorio al ingreso mostraron un hemograma sin datos patológicos. Fue valorado por el servicio de Ortopedia, que describe las radiografías de miembros inferiores mostrando hipomineralización generalizada, además, fractura en tercio medio de fémur derecho, tercio superior de fémur izquierdo, tercio superior de tibia derecha y tercio inferior de tibia izquierda, con deformidad curva, formación de callo óseo que ocupa la zona medular y cambios displásicos en metáfisis (Figura 2). A la vez se visualiza fractura de tercio medio de clavícula derecha y séptimo arco costal derecho, en el tercio medio con formación de callo óseo.
Por otro lado, el equipo de genética realiza el diagnóstico clínico de osteogénesis imperfecta (OI) tipo II con pronóstico reservado; en ese momento los padres recibieron consejo genético porque se determinó que eran primos segundos. Los estudios complementarios realizados se describen a continuación; ecocardiograma sin cardiopatía estructural de ningún tipo, fondo de ojo con el nervio óptico, mácula y retina sanas; ultrasonido de cerebro, abdomen, riñones y vías urinarias, sin hallazgos patológicos. En vista de que el paciente cursó estable y los resultados de los estudios enviados fueron favorables, se decidió egresar y continuar manejo en consulta externa. En cuanto a su evolución clínica, tres meses después del egreso se le realizó valoración audiológica (que no mostró hallazgos patológicos) y nutricional, con implementación de una dieta adecuada a los requerimientos. También continuó control en el Servicio de Genética; a la edad de 3 meses y 26 días pesó 3,3Kg., con una talla de 46cm. y un perímetro cefálico de 37,5cm., por lo que se clasifica con retardo moderado en el crecimiento; para esa fecha lo describen activo, hidratado, con las fontanelas amplias y diástesis de suturas, sin fracturas nuevas, con dos hernias -una inguinal derecha pequeña y otra umbilical-, ambas reducibles; el resto del examen físico no muestra alteraciones. Anotan que por el momento no se administra ningún fármaco y se tramita lo pertinente para adquirir el pamidronato.
Discusión
OI es un conjunto de trastornos genéticos que afectan la integridad del tejido conectivo, de ahí que los pacientes desarrollan fragilidad ósea ante traumas leves, deformidades esqueléticas progresivas, estatura baja, laxitud articular, adelgazamiento de la piel, pobre desarrollo muscular, dentinogénesis inadecuada y pérdida de la capacidad auditiva, en términos generales.1 Mediante estudios bioquímicos y de genética molecular se logró determinar que la mayoría de los individuos afectados portan mutaciones en los genes que codifican las cadenas polipeptídicas alfa1 y alfa 2 del protocolágeno tipo I, principal proteína de la matriz extracelular del tejido óseo, piel y tendones.2
Se estima que la incidencia de OI varía entre 1 en 10 000 (1, 2) y 1 en 20 000 nacidos vivos,3, 4 sin preferencia racial o étnica. Se reporta que en países occidentales la incidencia llega a ser hasta de 1 en 30 000.4 El diagnóstico de esta patología se fundamenta en hallazgos clínicos y radiológicos; se describe que en las formas leves donde no se identifican antecedentes familiares y el cuadro clínico evidencia fracturas no traumáticas, es necesario descartar otras entidades como osteoporosis idiopática y síndrome de niño agredido.5 Otros recursos empleados para el diagnóstico son la densidad mineral ósea, marcadores bioquímicos de reabsorción ósea, y estudios genéticos; sin embargo, estos estudios no siempre se encuentran al alcance. El tratamiento actual incluye intervenciones quirúrgicas por medio de estabilización intramedular con la colocación de prótesis, así como terapia farmacológica con calcio, fluoruros y calcitonina, que por ser agentes con resultados clínicos no muy favorables, se encuentran en desuso. Se debe aclarar que existen otros agentes terapéuticos como la hormona de crecimiento (HC) y los bisfosfonatos. Por su parte, el uso de HC estimula el metabolismo óseo e incrementa el crecimiento ponderal, factores que están disminuidos en las formas moderadas de la enfermedad. Sin embargo, estos beneficios no son evidentes en trastornos severos. Más recientemente se investiga el uso de bifosfonatos, terapia génica y transplante de células madre.6
La OI se divide en cuatro grupos de los cuales es fundamental describir sus características clínicas y diferenciación, para relacionar los hallazgos descritos en la bibliografía y el caso en estudio. (Cuadro 1)

En primer plano los casos de OI tipo I se manifiestan con fracturas al nacer únicamente en un 8% de los casos; hasta un 23% presenta fracturas en el primer año de vida, un 45% en la edad preescolar, y un 17% en la escolar. También se menciona que las deformidades en las extremidades son moderadas, ejemplo de estas serían la curvatura anterior y lateral del hueso femoral, o la curvatura anterior de la tibia, las cuales clásicamente inician en el periodo postnatal; además, es común que se desarrolle escoliosis o sifosis y pérdida auditiva hasta en un 50% de los pacientes.8 Este panorama se aleja del caso en estudio, ya que está claro que el paciente justo al nacer presentaba múltiples fracturas y curvatura con deformidad de las extremidades; no se documenta hiperextensibilidad articular ni deformidad en la columna vertebral, y durante la evolución se evidencia que la capacidad auditiva es normal.
Por otro lado, se logra determinar que el caso en estudio comparte una serie de hallazgos clínicos con los pacientes portadores de OI tipo II, ya que se describe que en estos hay una deficiencia en el crecimiento prenatal, con evidencia de fracturas múltiples en útero, las cuales consolidan con formación de callo óseo evidente en el periodo postranatal. En este caso se describe que el paciente presentaba fracturas en la clavícula derecha, séptimo arco costal, ambos fémur y tibias, datos que coinciden con este tipo de osteogenesis. Por su parte, los pacientes con OI tipo II presentan extremidades cortas y con deformidad angular, fontanelas amplias, escleras azules y hernias inguinales, como el caso en estudio.1,6
Respecto a lo que se menciona en la bibliografía sobre la OI tipo III, hay hallazgos que se comparten con el caso, como el déficit del crecimiento prenatal con fracturas múltiples presentes al nacimiento y deformidad ósea progresiva; sin embargo, estos pacientes tienen macrocefalia con la cara triangular, escleras no azuladas, pérdida auditiva y sifosis severa con compromiso respiratorio; características que el paciente del caso en estudio no presentaba. También se describe que el grado de fragilidad ósea es severo, y como se mostró en la evolución del paciente evaluado con tres meses de edad, este no había presentado durante todo el periodo, datos clínicos sugestivos de fracturas, por lo cual no correlacionaría con esta forma de OI.1
Respecto al tipo IV, no se describe que las fracturas y la deformidad en huesos largos estén presentes desde el nacimiento; además, difiere del caso en estudio porque estos pacientes suelen tener las escleras normales, intolerancia el calor, sudoración excesiva y apnea del sueño.
Dado que el diagnóstico de esta entidad es clínico y radiológico, es necesario profundizar en los hallazgos radiológicos, pues son fundamentales para descartar otras patologías.
En la bibliografía se menciona que en pacientes portadores de la enfermedad severa congénita, como el caso en estudio, los huesos largos de las extremidades son cortos, con las corticales delgadas, y la diáfisis es tan ancha como la metáfisis. También se describe que los niños presentan numerosas fracturas en útero o recientes, por lo cual se evidencian diversos estados de resolución del trazo de fractura, con una deficiente resolución de dichas fracturas, lo que predispone a que los pacientes desarrollen deformidades angulares en huesos largos, como la que se presenta en el caso en estudio.9
Por otra parte, se menciona que existen tres patrones radiológicos predominantes, el primero se caracteriza por presentar una densidad ósea aumentada, el segundo se describe como frágil, y el tercero es quístico.9
En el primer tipo el aspecto denso en el hueso está dado por la formación de callo óseo en las zonas de consolidación de las fracturas, que además asocia una cavidad medular estrecha y, en algunos casos, completamente obliterada. Estos hallazgos se pueden correlacionar con el caso en estudio, ya que la formación de callo óseo e invasión de la médula ósea por este, predominaba en las diversas fracturas del paciente.
En el segundo patrón se describe que las corticales y trabéculas de la cavidad medular son delgadas, con osteopenia marcada. Por último, el patrón quístico tiene un aspecto en "panal de abeja" en los huesos largos. Se ha descrito que pacientes que presentan crecimiento ponderal, pero que no caminan, tienen mayor riesgo de desarrollar este patrón. Otra variante de este patrón consiste en las calcificaciones con aspecto similar a "palomitas de maíz" (colecciones redondeadas radiolúcidas con margen esclerótico y opacidad central, dispuestas en racimos), tanto en metáfisis como en epífisis cerca de la placa de crecimiento en huesos largos. Se cree que posiblemente se desarrollan por fragmentación de la placa de crecimiento, ya que se ha visto que aumentan en pacientes con crecimiento acelerado, mientras que resuelven después de completar el crecimiento. Con estos hallazgos se puede afirmar que ninguno de los dos últimos patrones es compatible con los del paciente en estudio.
Cabe mencionar que en la mayoría de los casos severos de la enfermedad, existe osteoporosis marcada en la columna vertebral, que provoca compresión de los cuerpos vertebrales y desarrollo secundario de escoliosis o sifosis; sin embargo, estas alteraciones no se describen en el inicio ni durante la evolución del paciente en estudio.
Por su parte, en las formas más leves predomina la osteoporosis, que muestra la cortical delgada y el hueso trabecular del canal medular de aspecto delgado y difuminado. Las radiografías indican fracturas en diversos estadios de resolución, pero a diferencia de los casos severos, hay un remodelado adecuado del trazo de fractura. Es característico que los pacientes muestren una extremidad inferior con desviación el valgo y la otra el varo, con dislocaciones de las articulaciones patelofemoral, de cabeza del radio o de la cadera, asociando grados variables de escoliosis y sifosis. En vista de que las manifestaciones clínicas del paciente no coinciden con estas alteraciones leves, se cataloga como un caso de OI tipo II severa con patrón denso, predominantemente.9
En estos pacientes es necesario hacer diagnóstico diferencial con otras patologías; en primer plano se describe la hipofosfatemia congénita, que es una enfermedad letal, donde los hallazgos bioquímicos son esenciales para hacer diagnóstico; presenta un nivel sérico de fosfato disminuido, con pérdida de la actividad de la fosfatasa alcalina de los leucocitos, con excreción excesiva de fosforiletanolamida. Por otra parte, en neonatos la OI se confunde frecuentemente con acondroplasia; en este caso, al paciente lo refieren de Cartago con ese diagnóstico, sin embargo, los hallazgos radiológicos expuestos son fundamentales para descartar esta entidad. Otra patología por descartar sería la osteoporosis idiopática juvenil, caracterizada por ser autolimitada e iniciar justo antes de la pubertad en la mayoría de los casos, criterios que se alejan del caso en estudio. 3
En cuanto al manejo de esta patología hay que considerar que el objetivo fundamental es proporcionar la mayor funcionalidad posible, tomando en cuenta que el nivel de función alcanzable depende de la severidad de la enfermedad y de la edad del paciente. Así, por ejemplo, conforme el niño crece se ha observado que la tasa de fracturas disminuye progresivamente, tal y como se muestra en la evolución de este paciente, pues mejora la coordinación e incrementa la resistencia ósea. Sin embargo, con el crecimiento se desarrollan otras alteraciones, como la escoliosis, que amerita manejo ortopédico. De ahí que el manejo debe ser multidisciplinario, por parte de neonatología, genética, ortopedia, fisiatría, audiología, nutrición y soporte psicológico, para alcanzar este objetivo.
Antes de discutir sobre las nuevas perspectivas de tratamiento, es fundamental constatar que en la fisiopatología de la enfermedad se presenta un incremento en la actividad osteoclástica, por consiguiente reabsorción ósea y reducción de la síntesis ósea, porque justo en esta vía es que intervienen los bifosfonatos, al ser potentes antirresortivos. Se han desarrollado estudios científicos que implementan el uso de bifosfonatos en pacientes con OI severa, que presentan baja estatura y deformidad ósea marcada. Se demostró que el uso de pamindronato intravenoso intermitente disminuyó la reabsorción ósea, incrementó la densidad mineral ósea y redujo la tasa de fracturas; además, se documentó un efecto dramático en el bienestar y disminución del dolor óseo, que estimuló la deambulación. Pero, es necesario realizar más estudios para monitorear los efectos de estos fármacos en el largo plazo.5
Un nuevo enfoque en el tratamiento de esta enfermedad consiste en el uso de células madre, que potencialmente se Costa Rica en el exterior diferencien en osteoblastos maduros para sintetizar matriz ósea normal. Se menciona el reporte de un estudio de tres pacientes con OI severa quienes recibieron transplante de medula ósea y mostraron resultados favorables, al incrementar la densidad mineral ósea y disminuir la frecuencia de fracturas en dos de los pacientes, valorados tres meses después del transplante; sin embargo, se menciona que es preciso efectuar estudios más grandes y con seguimiento de estos pacientes en el largo plazo. La terapia génica constituye un desafío en el manejo de estos pacientes, de ahí que sea motivo de investigación rigurosa.2
Recibido: 15 de mayo de 2008 Aceptado: 17 de febrero de 2009
Referencias
1. Hoffman, Jodi D, Estrella E.Newborn. Presentation of connective tissue disorders. NeoReviews. 2007; 8: 110-119. [ Links ]
2. Niyibizi C, Smith P, Zhibao M, Robbins P, Evans C. Potential of gene terapy for treating osteogenesis imperfecta. Clin Otrthop and Relat Reser, 2000; 379: 126-133. [ Links ]
3. Mihran O, Tachdjlan M. Pediatric Orthopedics. 1990; 2 ed Saunders Company; Vol II: 1-3366. [ Links ]
4. Paterson Colin R, McAllion Susan J. Classical Osteogenesis Imperfecta and Allegations of Nonaccidental Injury Clin Orthop and Relat Reser, 2006; 452: 260-264 [ Links ]
5. Devogelaer J. New uses of biphosphonates: osteogenmesis imperfecta. Current Opinion in Pharmacol. 2002; 2:748-753. [ Links ]
6. Peter H. Byers. Osteogenesis imperfecta: perspectives and opportunities. Current Opinion in Pediatrics, 2000;12:603-609. [ Links ]
7. Astrom E, Soderhall S. Beneficial effect of biphosphonate during five years of treatment of severe osteogenesis imperfecta. Acta Paedriatr 1998;87:64-68 [ Links ]
8. Jones, K. Smit´s. Recognizable Patterns of Human Malformation. 1997; 5 ed. Saunders Company. Páginas 123-125. [ Links ]
9. Hanscom DA, Winter RB, Lutter L, Lonstein JE. Osteogenesis imperfecta: Radiographic classification, natural history, and treatment. J Bone Joint Surg Am. 1992; 74:598-616. [ Links ]
1 Médico Pediatra, Residente de Neonatología, Hospital Nacional de Niños
2 Bachiller en Ciencias Médicas y Licenciada en Cirugía y Medicina, Universidad de Costa Rica. Servicio de Neonatología, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera".
Abreviaturas: HC,Hormona de crecimiento;OI,osteogénesis imperfecta; RNTPEG, recién nacido de término pequeño para la edad gestacional.
Correspondencia: Gilberto Rodríguez Herrera. Email: gilbertorh@yahoo.com

