










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.50 n.1 San José Mar. 2008
Diagnóstico molecular de la enfermedad de Huntington en Costa Rica
Molecular diagnosis of Huntington´s disease in Costa Rica
Melissa Vásquez-Cerdas1,4, Fernando Morales-Montero1,4, Húbert Fernández-Morales2, Gerardo el Valle-Carazo3,Jaime Fornaguera-Trías4, Patricia Cuenca-Berger1,4
Resumen
Justificación y objetivo. Este estudio representa un esfuerzo para establecer por primera vez en Costa Rica el diagnóstico molecular de la enfermedad de Huntington; esto favorecerá un mejor manejo clínico de los pacientes y podrá ser traducido en un incremento de la calidad de vida de las familias. Se pretende determinar el número de repeticiones CAG en personas con la enfermedad de Huntington y familiares mediante el diagnóstico molecular,con el fin de ofrecerles asesoramiento genético decuado y oportuno.
Métodos:El estudio se realizó en 7 pacientes con diagnóstico clínico de Huntington y 31 familiares en riesgo. Para determinar el número de repeticiones CAG se utilizó la reacción en cadena e polimerasa a posterior electroforesis sobre geles de agarosa y poliacrilamida.
Resultados:Se obtuvo el diagnóstico molecular de los 38 individuos.Se confirmó el diagnóstico clínico en las 7 personas afectadas, se encontraron 11 con la mutación que permanecían asintomáticas y 20 sin la mutación. Se observó una correlación negativa entre la edad de inicio y el número de repeticiones, así como inestabilidad intergeneracional, tanto vía materna como paterna. No hay diferencias en el número de repeticiones, según el sexo del progenitor transmisor.
Conclusión: Los análisis moleculares mostraron un perfil de repeticiones similar al de otras poblaciones.Hemos identificado las primeras familias portadoras de enfermedad de Huntington en Costa Rica, permitiendo dar a los pacientes y su familia asesoramiento genético adecuado y oportuno basado en información confiable.
Descriptores: Costa Rica, diagnóstico molecular, enfermedad de Huntington, inestabilidad genética, mutaciones inestables, tripleta repetida CAG
Abstract
Justification and Aim.This study represents an effort to establish the molecular diagnosis of Huntington s disease in Costa Rica. This would improve the clinical management of the patients and that could be translated into better quality of life for them and their families. Aim: to determine the number of CAG repeats in affected individuals and their relatives by molecular diagnosis in order to offer them adequate genetic counseling.
Methods:The study involved 7 patients with clinical diagnosis of this disease and 31 relatives at risk. To determine the number of repeats we used the polymerase chain reaction and electrophoresis on agarose and polyacrylamide gels.
Results:We obtained the molecular diagnosis in all of the individuals. The clinical diagnosis was confirmed in the 7 affected individuals,11 remained asymptomatic but carrying the mutation and 20 were found without the mutation. A negative correlation was observed between the age of onset and the repeat size. We also observed intergenerational instability, both through the maternal and paternal side. There were no differences in the number of repeats according to sex of the progenitor transmitter.
Conclusions: Our molecular analyses showed a profile of repeats similar to other populations. We have identified for the first time families carrying a CAG expansion in Costa Rica,which will allow us to give adequate and opportune genetic counseling to the patients and their families, based on reliable information.
Key words: Costa Rica, molecular diagnosis, Huntington disease; genetic instability, instable mutations, AG repeat triplets.
Recibido: 1 de junio de 2007 Aceptado: 2 de septiembre de 2007
La enfermedad de Huntington (HD) es neurodegenerativa progresiva del sistema nervioso central, afecta áreas determinadas del cerebro,específicamente corteza cerebral y estriado (caudado y putamen). Se encuentra en todos los grupos étnicos y la prevalencia está estimada entre 5 y 10 personas afectadas por cada 100,000 habitantes, entre poblaciones de Europa Occidental y descendientes de estas poblaciones (poblaciones caucásicas).1,2
Las manifestaciones clínicas se caracterizan por un cuadro progresivo de movimientos anormales, rápidos e involuntarios de tipo coreico, que afectan con mayor frecuencia a los miembros inferiores y a la cara, además de trastornos siquiátricos eterioro rogresivo e irreversible de las funciones cognitivas. 3
La edad de manifestación de la HD es altamente variable y la incidencia en ambos sexos es similar. Por lo general, se presenta entre los 30 y 50 años de edad (HD clásica), cuando muchas personas ya se han reproducido y han formado sus familias, por eso se dice que es una enfermedad de manifestación tardía.4
Presenta un patrón de herencia autosómico dominante con expresividad variable y penetrancia completa: todos los individuos que hereden el alelo mutado eventualmente desarrollarán la enfermedad, a menos que mueran de otras causas antes del inicio de los síntomas. 5
El defecto génico consiste en una expansión del trinucleótido CAG (Citosina, Adenina, Guanina)6 cerca del extremo 5, en el exón 1 del gen inicialmente llamado IT15 (interesting transcript 15) y ahora llamado gen HD, localizado en el brazo corto del cromosoma 4 (4p16.3). Las expansiones son traducidas en un segmento de poliglutamina (poliQ) cerca del amino terminal, en la proteína que ha sido llamada huntingtina (htt).7
La expansión es inestable,lo que significa que el tamaño de la secuencia repetida varía cuando las células se dividen, tanto en la línea germinal como somática.8 Los individuos normales poseen de 6 a 35 repeticiones CAG. Los individuos con la D poseen más de 36 repeticiones CAG.9
La htt es una proteína de expresión ubicua. Se expresa altamente en cerebro y en una amplia variedad de tejidos (testículos, ovarios, pulmón, hígado e intestino), en todas las etapas del desarrollo (se sugiere que es importante en la maduración de neuronas) y en el adulto.10 El gen HD mutante se expresa en el cerebro en niveles comparables con los del gen HD normal desde los estadios tempranos del desarrollo embrionario. Dentro de las neuronas, la htt se encuentra fundamentalmente en el citoplasma, asociada con membranas o citoesqueleto en el soma o cuerpo celular, dendritas, axones y terminales nerviosas.11
Hay varios aspectos aún desconocidos acerca de la HD. Muchos de ellos están aún en debate entre la comunidad científica de todo el mundo,como son:el mecanismo responsable de la expansión y la vía que lleva a la neurodegeneración.
El objetivo del estudio consistió en determinar el tamaño exacto de la mutación en personas afectadas con la HD y sus familiares mediante el uso de técnicas de biología molecular, con el fin de conocer cómo se está expresando la mutación en las familias costarricenses y así poder ofrecerles el asesoramiento genético adecuado y oportuno.
A pesar de ser la HD una terrible enfermedad que demanda atención,no ha sido estudiada en Costa Rica ni se conoce su prevalencia.Este estudio representa un primer esfuerzo para establecer en el país el diagnóstico molecular para la HD e iniciar su investigación.Por ser una enfermedad neurodegenerativa,progresiva y totalmente discapacitante,es preciso conocer la población costarricense que porta la mutación,e implementar los métodos diagnósticos moleculares,ya que esto es requisito indispensable para ofrecer el consejo genético adecuado y oportuno.En última instancia,esto favorecerá un mejor manejo clínico de los pacientes y,por lo tanto,podrá traducirse en un incremento considerable de la calidad de vida de las familias con la HD.
Pacientes y métodos
Población de estudio
Pacientes con un diagnóstico clínico de HD realizado por un neurólogo, así como los familiares de estos pacientes con riesgo de presentar la mutación que causa la HD. La mayoría de los casos índices fueron localizados en el Hospital Nacional Psiquiátrico mediante la revisión de expedientes, un caso fue remitido por el Hospital Calderón Guardia y otro por el Laboratorio de Neurofisiología NeuroLab. El protocolo del estudio, así como la fórmula de consentimiento informado fueron aprobados por los comités éticos científicos de la Universidad de Costa Rica y del Hospital Nacional Psiquiátrico de la Caja Costarricense de Seguro Social. Todas las personas que accedieron a participar en el estudio firmaron la fórmula de consentimiento informado.
Diagnóstico Molecular
A cada persona se le tomó una muestra de sangre periférica con anticoagulante ACD (ácido cítrico,citrato de sodio y dextrosa) y se obtuvo el ácido desoxirribonucleico (ADN) de leucocitos mediante extracción con fenol-cloroformo,según los procedimientos usuales.12 Para determinar el número de las repeticiones CAG se realizó la reacción en cadena de la polimerasa (PCR).El protocolo de la PCR usado fue el descrito por Warner et al.13 ,con algunas modificaciones.Se amplificaron 150 ng de ADN genómico siguiendo las condiciones usuales,con buffer 10X,2U de Taq polimerasa, agua y 0.5 µM de cada uno de los iniciadores HD1 y HD3 , que amplifican selectivamente las repeticiones CAG.
La PCR se llevó a cabo en un volumen de reacción de 25 µL.El perfil de reacción utilizado en un termociclador Perkin Elmer (GGeneAmp PCR System 2400) fue el siguiente: una desnaturalización inicial del ADN por 4 min a 94 °C, seguida por 35 ciclos de 94 °Cpor 30S, 65 °Cpor 30S y 45S de extensión a 72 °C. Finalmente un período de extensión última 2°C por 10 min.
Después los productos de la PCR se corrieron en minigeles de agarosa al 1.5%, teñidos con bromuro de etidio, en buffer TBE 1X pH:7.7 durante 1 hora con el fin de confirmar si había producto amplificado.Luego,los productos de la PCR se sometieron a una electroforesis sobre geles de poliacrilamida desnaturalizante al 6% ó al 8%, en buffer TBE 1X pH: 8.0 por un período de 2h 50min, para determinar el tamaño absoluto de los fragmentos. Los fragmentos fueron visualizados mediante el método de tinción con nitrato plata (Figura 1). Los alelos son considerados normales cuando tienen menos de 36 repeticiones y expandidos cuando contienen más de 40 CAGs. Los alelos que tienen de 36 a 39 repeticiones presentan penetrancia incompleta.
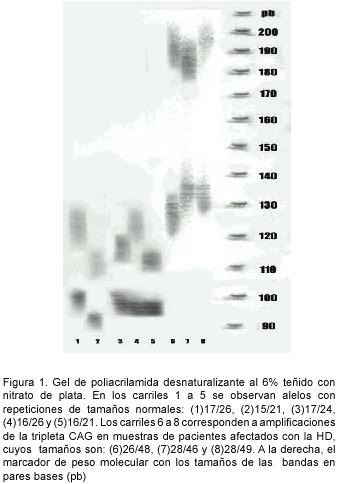
Asesoramiento y entrega de resultados
Los pacientes y sus familiares recibieron asesoría genética,así como evaluación y ayuda psicológica con el fin de garantizarles apoyo continuo durante el proceso de decidir si se sometían o no al diagnóstico molecular, si querían o no saber el resultado y, posteriormente, cuando conocieron si eran o no portadores del gen con la mutación.Una vez que se tenían los resultados moleculares, la información se ofrecía tanto de manera oral como por escrito. Algunos de los casos positivos para la mutación están siendo seguidos por los neurólogos y el psicólogo. Los resultados de este seguimiento serán publicados después.
Análisis estadístico
Para establecer la correlación entre el tamaño de la mutación y la edad de inicio,se utilizó el coeficiente de correlación r de Pearson (nivel de significancia al 1%).Para determinar si existen diferencias en cuanto al sexo afectado y la preferencia en la transmisión del alelo HD según el sexo del progenitor afectado se realizaron pruebas de Chi-cuadrado (nivel de significancia al 5%).Para determinar si existen diferencias en el tamaño de los alelos expandidos según el sexo afectado, y la transmisión (mmaterna o paterna), se realizó la prueba U de Mann-Whitney (nivel de significancia al 5%).
Resultados
El diagnóstico molecular se diagnosticó a 38 personas pertenecientes a 4 diferentes familias costarricenses. De estos individuos, 7 tenían un diagnóstico clínico de HD y los restantes 31 personas correspondían a familiares en riesgo. En total participaron 21 mujeres (55.33%) y 17 varones (44.77%).
Se logró confirmar el efectúo clínico de la HD en los casos índices y, por lo tanto, la presencia de la HD en las 4 familias estudiadas. En el caso de los 31 individuos en riesgo, la mayoría manifestaron su interés por conocer si son o no ortadores de a mutación y la posibilidad de heredarla a sus hijos.
Las expansiones (mutaciones) fueron observadas en 18 individuos. Las características moleculares de estos 18 individuos positivos para la mutación se pueden observar en el Cuadro 1.
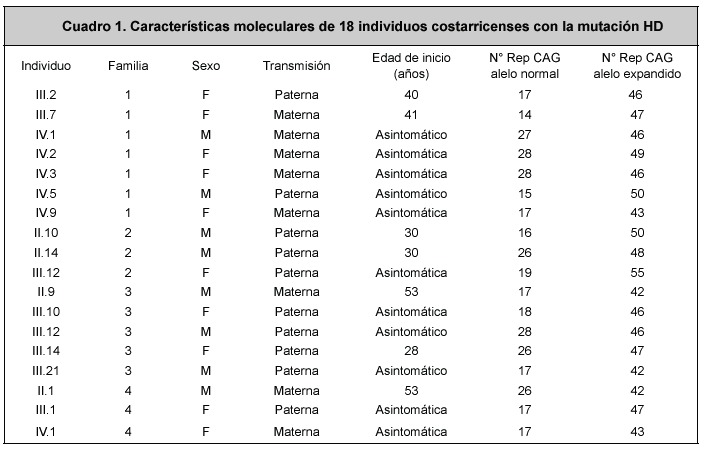
De 18 individuos con la mutación (positivos), un 44.4% eran hombres y un 55.6% mujeres, por lo que no hay diferencias significativas relacionadas con el sexo afectado (X2 = 0.22, P=0.05, gl=11). La relación entre individuos afectados (n=7) sintomáticos n=11) es 1:1.6.
Todos los 18 pacientes en los que se encontró la mutación resultaron ser heterocigotas, con un alelo normal y un alelo expandido. El número de repeticiones CAG en los alelos mutados varió de 42 a 55, con un promedio de 46.4 repeticiones CAG (SSD ±3.44), siendo el alelo más común de 46 repeticiones (27.8%). Tomando en cuenta los alelos normales en los individuos en los que se detectó la mutación (n=18), estos presentaron entre 14 y 28 repeticiones, con un promedio de 20.7 repeticiones CAG (SSD ±5.33) y un alelo modal de 7 repeticiones (38.9%).
No se encontraron diferencias significativas en el tamaño de los alelos expandidos entre hombres afectados (promedio= 45.75 CAGs) y mujeres afectadas (promedio=46.9 CAGs)(U Mann Whitney,P>0.05).
Se encontró además que 10 de los alelos expandidos fueron transmitidos vía paterna y 8 de los alelos expandidos fueron transmitidos vía materna (Cuadro 1), de manera que no hay diferencias significativas en la transmisión del alelo expandido, según el sexo del progenitor transmisor (X2= 0.22, P=0.05, gl=1). Por otra parte, no se encontraron diferencias significativas en el tamaño de las repeticiones CAG expandidas entre pacientes en los que la transmisión fue paterna (promedio= 47.7 CAGs) y aquellos con transmisión materna (promedio=44.7 CAGs)(U Mann Whitney, P>0.05). No se encontraron alelos intermedios entre 36 y 39 repeticiones en este grupo.
En las 4 familias se observó inestabilidad de la repetición CAG durante la transmisión de una generación a la siguiente, tanto vía materna como paterna. La mutación fue inestable en el 62.5% (5/8) de las transmisiones intergeneracionales para las que se tienen los datos. En general, los cambios son pequeños, de solamente unas pocas repeticiones CAG (entre 3 y 5 repeticiones), con incrementos y disminuciones. En las restantes 3 parejas progenitor-hijo(a) no se observaron cambios en el número de repeticiones (CAG Cuadro 2).
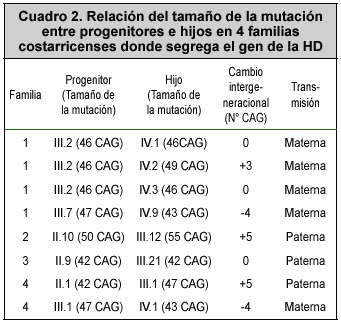
A pesar de que la magnitud de los cambios observados fue pequeña, hay una tendencia hacia la expansión o contracción de las repeticiones, determinada por el sexo del progenitor que está transmitiendo la HD. Cuando se compara el tamaño de la mutación en los progenitores y en los hijos, se observa que en los casos cuando la transmisión es vía materna, con la excepción de uno, se transmite un alelo de igual o menor tamaño (contracciones en el número de repeticiones), mientras que por el contrario, cuando la transmisión es paterna, el alelo con la mutación en el hijo es de mayor tamaño que el alelo con la mutación en el padre, excepto en un caso donde se hereda un alelo con el mismo número de repeticiones (Cuadro 2). Es importante destacar que en el individuo con 55 repeticiones, la mayor expansión de la tripleta CAG encontrada, la transmisión fue por vía paterna.
En este estudio, la edad de inicio de la enfermedad se basa en la información provista por el paciente o un familiar cercano. La edad promedio de inicio para la población en estudio fue de 39.3±10.6 años (ámbito: 28-53 años) y la mayoría desarrollaron la HD en la cuarta década de vida.
Con respecto a la edad de inicio, debe destacarse el caso de las pacientes III.7 de la familia #11 y la III.14 de la familia #3, ya que ambas presentan el mismo número de repeticiones (47 CAG) en el alelo expandido, sin embargo, hay una gran diferencia en la edad de inicio.
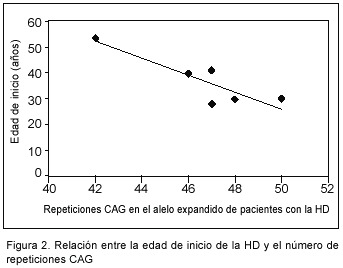
Cuando se analizan los casos en conjunto, no por generaciones, se puede observar que la edad de inicio está relacionada estadísticamente con el número de repeticiones CAG en el alelo expandido (r= -00.914, P<0.01) (Figura 2). La correlación es negativa y esto significa que cuanto mayor sea el número de repeticiones, más temprano aparece la enfermedad.
Discusión
Este es el primer estudio que se realiza en Costa Rica sobre la enfermedad de Huntington,en el cual se identifican las primeras 4 familias portadoras de la mutación en el gen de la HD en el país. Este esfuerzo, junto con la colaboración de los profesionales de la salud, permitirá definir políticas para establecer en un futuro la prevalencia de este mal. Sin embargo, es de esperar que la prevalencia de la HD en la población costarricense sea similar a la observada en caucásicos, a que, e acuerdo con algunas investigaciones, el genotipo promedio de la población costarricense está conformado por una mezcla acumulada de genes esencialmente de origen caucásico (61%), seguido por el aporte amerindio (30%) y el africano (9%).14
Los análisis moleculares mostraron una distribución del número de repeticiones CAG dentro de intervalos previamente reportados en otras poblaciones a nivel mundial.15-22 Los alelos normales más comunes en el mundo poseen 17 y 19 repeticiones CAG;9 en el estudio este alelo normal más frecuente posee 17 repeticiones. Por otro lado no es sorprendente que no se hayan encontrado alelos intermedios, ya que las repeticiones en este intervalo son raras. Aproximadamente, del 1.5% al 2% de la población general lleva alelos en este intervalo, asimismo, los alelos de penetrancia reducida también son raros en la población.9
En la gran mayoría de las transmisiones, tanto maternas como paternas, la repetición expandida (aaproximadamente entre 40 y 50 repeticiones) es inestable, incrementando o disminuyendo levemente en una o pocas repeticiones CAG de una generación a la siguiente.23
Aunque la inestabilidad es independiente de la herencia materna o paterna, Kremer et al .24 encontraron que las expansiones grandes son casi exclusivas de las transmisiones paternas. Aproximadamente el 21% de las transmisiones paternas están asociadas con expansiones grandes. En contraste, las expansiones solo son observadas en el 0.7% de las transmisiones maternas y las contracciones ocurren más frecuentemente en los alelos derivados de la madre. Las expansiones de las repeticiones CAG vía materna, han sido reportadas solo en el 20% de los casos juveniles de HD y raramente sobrepasan las 20 repeticiones.25 Cannella et al.26 reportaron un promedio de expansión intergeneracional de +7.3 repeticiones en las transmisiones paternas y de +0.7 repeticiones en las maternas.
El sexo del progenitor transmisor es uno de los elementos más importantes que influyen en la estabilidad de los alelos HD. Varios estudios han mostrado que los alelos CAG expandidos son particularmente inestables durante la transmisión a través de la línea germinal masculina,23 y es más probable que las transmisiones paternas produzcan grandes expansiones causantes de la HD juvenil. En el estudio no se presentó ningún caso de HD juvenil y tampoco se observaron expansiones grandes o dramáticas en las transmisiones paternas,lo cual pudo influir en el hecho de que no se presenten diferencias significativas en el tamaño de las repeticiones CAG expandidas entre pacientes en los que la transmisión fue paterna y aquellos con transmisión materna.27,28 Se señala a la gametogénesis como la fuente principal de la inestabilidad, sin embargo, se desconoce el mecanismo molecular exacto que causa grandes expansiones durante la gametogénesis en los hombres. 29
El factor fundamental que influencia la edad de inicio en la enfermedad de Huntington es el número de repeticiones CAG. Varios estudios han mostrado que el número de repeticiones del trinucleótido CAG correlaciona de forma inversa con la edad a la cual la enfermedad comienza a manifestarse.7,9,22,27 Las expansiones entre 40 y 50 repeticiones CAGs son vistas con frecuencia en personas en quienes la enfermedad se presenta entre los 30 y los 50 años, como en estos casos, mientras que la HD de inicio juvenil se manifiesta,por lo general,cuando se sobrepasan las 70 repeticiones.30
Las expansiones en los alelos mutados encontrados en este estudio están entre las 42 y 55 repeticiones CAG, sin embargo, a pesar de que son muchos los estudios en los que se ha visto que la correlación entre la edad de inicio y el tamaño de la expansión es fuerte, gran parte del efecto es observado entre la pequeña fracción de individuos que tienen muchas repeticiones (=70 CAGs)y edad de inicio temprano o juvenil (10%). En el 90% de los casos donde la edad de inicio se presenta en la vida adulta, la correlación entre el tamaño de la expansión y la edad de inicio no es lo suficientemente fuerte y significativa para ser útil en la predicción del inicio de la enfermedad.7
Según lo mencionado, es de esperar que en el grupo estudiado se observe una correlación débil entre la edad de inicio y el número de repeticiones CAG, pero a correlación negativa es fuerte (r=-0.914) (Figura 2), lo que probablemente se deba a que para este análisis se contaba con una muestra muy pequeña, ya que solo 7 pacientes habían desarrollado la enfermedad y fue en ellos que se pudo obtener la edad de inicio de los síntomas.
El incremento en la cantidad de las tripletas CAG que se observa entre generaciones, en algunas familias, provee la base molecular para el fenómeno de anticipación, que se explica por la inestabilidad en la vía germinal que exhiben las repeticiones CAG expandidas. En la HD se produce, por lo general, cuando la transmisión de la mutación es por vía paterna.7 En estos casos, mediante el diagnóstico molecular se observó que en 3 de las transmisiones intergeneracionales (2 vía paterna y 1 vía materna) un progenitor afectado heredó a su hijo un alelo mutado expandido (Cuadro 2). Por el momento, estos hijos permanecen asintomáticos, pero a pesar de que ninguno ha alcanzado ni sobrepasado la edad de inicio de la HD en sus respectivos progenitores, es posible que en estos casos no se observe una clara anticipación, ya que el incremento en il número de repeticiones es pequeño.
Muchos estudios han mostrado que hay una gran diferencia en la edad de inicio de la HD en diferentes familias, incluso entre individuos con idéntica longitud de repeticiones en el gen HD, como el caso reportado en este estudio (paciente III.7 de la familia #1 y la paciente III.14 de la familia #3) (Cuadro 1), y han referido que esta diferencia es atribuible a modificadores genéticos y ambientales que actúan en conjunto con el gen de la HD.22 Aunque también es preciso recalcar la dificultad para establecer la edad en que los pacientes manifestaron sus primeros síntomas. Este caso no solamente demuestra el pobre poder predictivo para la edad de inicio que tiene el número de repeticiones CAG, sino también la variación sustancial en la edad de inicio que no s explicada por el tamaño de la mutación.
La identificación de los factores modificadores,en especial aquellos que llevan a posponer la edad de inicio mucho más tarde en la vida y a prolongar los años productivos, es algo que podría llegar a ser muy ventajoso en la búsqueda y desarrollo de nuevos agentes terapéuticos que puedan estar disponibles y accesibles a todos.22
Los datos obtenidos en este primer estudio de la HD en Costa Rica coinciden con los hallazgos en otras partes del mundo, es decir, la mutación que causa la HD en las familias costarricenses tiene el mismo comportamiento: número de repeticiones CAG similar a los reportados en otras poblaciones en el nivel mundial, tendencia hacia la expansión o contracción de las repeticiones determinada por el sexo del progenitor que está transmitiendo la enfermedad y número de repeticiones CAG que muestra una correlación negativa con la edad de inicio de la HD.
Dado que por el momento los tratamientos se dirigen a reducir la sintomatología, y mientras no se pueda "curar " la condición de portador de la mutación, la prevención primaria de la ocurrencia de esta patología se basa en la asesoría genética adecuada y oportuna a los miembros de las familias afectadas.
Agradecimientos y colaboradores
La Vicerrectoría de Investigación de la Universidad de Costa Rica contribuyó a financiar este proyecto,bajo el N ° 742-A4-3304, así como también el Consejo Nacional para Investigaciones Científicas y Tecnológicas (CONICIT)de Costa Rica, a través de su programa FORINVES. El Organismo Internacional de Energía Atómica (OIEA) y el Servicio Alemán de Intercambio Académico (DAAD) han facilitado la adquisición del equipo adecuado para el estudio de nfermedades causadas por mutaciones inestables.
Referencias
1. Harper PS. The epidemiology of Huntington s disease. Hum Genet. 1992; 889: 3365-76. [ Links ]
2. Burguera JA, Solís P, Salazar A. Estimación de la prevalencia de la enfermedad de Huntington por el método captura-recaptura en la comunidad valenciana. eurología. 97;25:1845-77. [ Links ]
3. Deus-Y J, Pujol J, Espert R. Deterioro neurosicológico en la enfermedad e Huntington. eurología. 997;25:1257-68. [ Links ]
4. Myers RH, Vonsattel JP, Stevens TJ, Cupples LA, Richardson EP, Martin JB, et al. Clinical and Neuropathological assessment of severity in Huntingtons disease. Neurology. 1988;38:341-47. [ Links ]
5. Gusella J, Young AB. Huntington s Disease. En Conneally M, ed. Molecular Basis of Neurology.Boston:Blackwell Scientific Publications. 1993:113-127. [ Links ]
6. The Huntington`ss Disease Research Collaborative Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on untingtons disease chromosomes. Cell. 1993;72:971-83. [ Links ]
7. Myers RH, Marans KS, MacDonald ME. Huntington s disease. En Wells RD, ST Warren, eds. Genetic Instabilities and Hereditary Neurological iseases. alifornia: Academic. 1999:301-23. [ Links ]
8. Cuenca P, Morales F. Mutaciones inestables: causa de algunas enfermedades neurológicas hereditarias. Acta Méd Costarric. 1999;41:77-15. [ Links ]
9. Potter NT, Spector EB, Prior TW. Technical Standards and Guidelines for Huntington isease Testing. Genet Med. 2004;6:61-55. [ Links ]
10. Schilling G, Sharp A, Loev S, Wagster M, Hua-Li S, Colin O, et al. Expression of the Huntington s disease (IIT15) protein product in HD patients. Hum Mol Genet. 1995;4:1365-71. [ Links ]
11. Martín-Aparicio E, Lucas JJ. Bases moleculares de la enfermedad de Huntington y posibles mecanismos patogénicos. Neurología. 2002;35:212-20. [ Links ]
12.Straus WM. Preparation of genomic DNA from mammalian tissue. En Ausubel FM, Brent R, Kinston RE, eds. Current protocols in molecular biology. New York: Wiley. 1988:2.2.1-2.2.3 [ Links ]
13.Warner JP, Barron LH, Brock DJH. A new polymerase chain reaction (PCR) assay for the trinucleotide repeat that is unstable and expanded on Huntington s disease chromosome. Mol Cell Probes. 1993;77:235-39. [ Links ]
14. Morera B, Barrantes R, Marín-Rojas R. Gene Admixture in the Costa Rican Population. Ann Hum Genet. 2003;67:71-80. [ Links ]
15.Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington ´sdisease. Nat Genet. 1993;44:398-403. [ Links ]
16. Whitefield JE,Williams L,Snow K,Dixon J,Winship I,Stapleton PM, et al. Molecular analysis of the Huntington ´sdisease gene in New Zealand. NZ Med . 1996;109:27-30. [ Links ]
17.Sánchez A, Castellvi-Bel S, Mila M, Genis D, Calopa M, Jiménez D, et al.Huntington ´s disease:confirmation of diagnosis and presymptomatic testing in spanish families by genetic analysis. J Neurol Neurosurg Psychiatry. 1996;61:625-7. [ Links ]
18.Alonso ME, Yescas P, Cisneros B, Martínez C, Silva G, Ochoa A, et al. Analysis of the (CAG)n repeat causing Huntington ´sdisease in a Mexican population. Clin Genet. 1997;51:225-30. [ Links ]
19.Lima e Silva T, Guerra H, Bertuzzo C, Lopes I. Molecular diagnosis of Huntington disease in Brazilian patients. Arq Neuropsiquiatr. 2000;58:11 [ Links ]
20. Saleem Q,Roy S,Murgood U,Saxena R,Verma IC,Anand A,et al. Molecular analysis of Huntington ´sdisease and linked polymorphisms in the Indian population. Acta Neurol Scand. 2003;108:281-86. [ Links ]
21.Akbas F,Erginel-Unaltuna N.DNA testing for Huntington disease in the Turkish opulation. Eur Neurol. 2003;50:20-44. [ Links ]
22.Wexler N,The U.S.–Venezuela Collaborative Research Project. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington s disease age of onset. Proc Nat Acad Sci. 2004;101:498-503. [ Links ]
23.Laccone F, Christian W. A recurrent expansion of a maternal allele with 36 CAG repeats causes Huntington Disease in two sisters. Am J Hum Genet. 2000;66:1145-48 [ Links ]
24.Kremer B, Almqvist E, Theilmann J, Spence N, Telenius H, Goldberg YP, et al. Sex-dependent mechanisms for expansions and contractions of the CAG repeat on affected Huntington disease chromosomes. Am J Hum Genet. 1995;57:343-50. [ Links ]
25.Sánchez A, Mila M, Castellvi-Bel S, Rosich M, Jiménez D, Badenas C, et al. Maternal transmission in sporadic Huntington ´sdisease. J Neurol Neurosurg Psychiatry. 1997;62:535-7. [ Links ]
26.Cannella M, Gellera C, Maglione V, Giallonardo P, Cislaghi G, Muglia M, et al. The gender effect in juvenile Huntington disease patients of Italian origin. Am J Med Genet B Neuropsychiatr Genet. 2004;1125:92-98. [ Links ]
27. Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, et al. Trinucleotide repeat length instability and age of onset in Huntington ´s disease. . Nat Genet. 1993;4:387-92. [ Links ]
28.Telenius H, Almqvist E, Kremer B, Spencer N, Squitieri F, Nichol K, et al. Somatic mosaicism in sperm is associated with intergenerational (CAG)n changes in Huntington disease. Hum Mol Genet. 1995;44:189-95. [ Links ]
29. Bates GP. The molecular genetics of Huntington disease-a history. Nat Rev Genet. 2005;6:766-73. [ Links ]
30.Petersén A, Mani K, Brundin P. Recent Advances on the Pathogenesis of Huntington s Disease.Exp Neurol.1999;157:1 –18. [ Links ]
1. Instituto de Investigaciones en Salud (INISA),UCR.
2. Servicio de Neurología- Hospital Dr.Rafael A. Calderón Guardia
3. Laboratorio de Neurofisiología Neurolab
4. Programa de Investigación en Neurociencias, Escuela de Medicina, UCR
Abreviaturas: ADN, ácido desoxirribonucleico; CAG, Citosina Adenina Guanina; HD, enfermedad de Huntington; Htt, proteína huntingtina; PCR, reacción en cadena de la polimerasa.
Correspondencia: M.Sc. Melissa Vásquez Cerdas. Código postal:2060 San José. Tel.207-3050.Correo electrónico: melissav@cariari.ucr.ac.cr

