










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.49 n.3 San José Jul. 2007
Arteritis de Takayasu de evolución fulminante en una paciente pediátrica
Maikel Vargas-Sanabria, Mayela Valerio-Hernández
Departamento de Medicina Legal, Poder Judicial.
Correspondencia: Maikel Vargas Sanabria mvargassa@poder-judicial.go.cr Mayela Valerio Hernández mvalerio@poder-judicial.go.cr
Resumen
Se presenta el caso de una lactante de 4 meses, conocida sana, con un cuadro de 4 días de evolución, inicialmente inespecífico, que el último día se tornó fulminante, con signos francos de hipoperfusión tisular, que la llevaron a 2 paros cardiorrespiratorios y a la muerte. Al realizarse la autopsia médico legal y el estudio histopatológico se encontró una vasculitis de grandes vasos, correspondiente con enfermedad de Takayasu. La arteritis de Takayasu es por sí misma muy poco frecuente en el hemisferio occidental, esto aunado a que se manifieste en una lactante caucásica con evolución fulminante, constituye una entidad extremadamente rara y por lo tanto de interés para un diagnóstico más expedito.
Descriptores: vasculitis de la infancia, arteritis de Takayasu, insuficiencia cardiaca congestiva, arteritis de células gigantes, estenosis y obliteración arterial.
Key words: childhood vasculitis, Takayasu arteritis, congestive heart failure, giant cells arteritis, arterial stenosis.
Recibido: 23 de agosto de 2006 Aceptado: 27 de febrero de 2007
Caso
Se trata de una paciente de 4 meses de edad, conocida sana, quien a principios de mayo de 2006 fue llevada al centro de salud de su localidad por un cuadro de tos seca de 4 días de evolución, sin fiebre ni dificultad respiratoria. Allí se le prescribió prednisolona líquida y se continuó manejo ambulatorio, sin embargo, después de administrarle la primera dosis de este medicamento, presentó vómitos de contenido gástrico, flacidez, desviación de la mirada y cianosis peribucal. Esto ameritó su traslado a una clínica periférica, donde fue estabilizada y enviada al centro de atención de tercer nivel (HNN). Allí, desde su ingreso, mostró una condición crítica con signos como taquipnea con quejido y tirajes intercostales (++), palidez, llenado capilar lento y roncus pulmonares bilaterales. Evolucionó tórpidamente e hizo un paro cardiorespiratorio (PCR) inicial del cual se logró rescatar después de más de 20 minutos de maniobras. A pesar de esta recuperación continuó deteriorándose, presentando francos datos de hipoperfusión tisular: taquicardia, acidosis metabólica grave y oliguria. Se iniciaron esteroides en dosis de insuficiencia suprarrenal, pero, pocas horas después presentó otro PCR del cual no se logró recuperar y falleció.
En la autopsia médico legal, el hallazgo más significativo fue el engrosamiento y endurecimiento de la paredes de los grandes vasos, con la consecuente estenosis luminal y en algunos segmentos luz puntiforme, en sitios como aorta, carótidas, arterias pulmonares y coronarias (Figuras 1a y 1b).
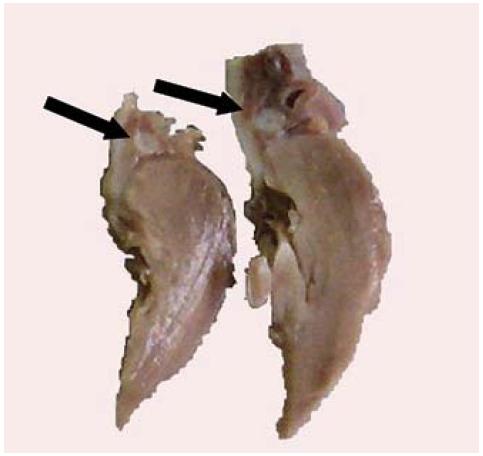
Figura 1a: Corte transversal de arteria coronaria. Las flechas señalan el engrosamiento de las paredes y la marcada estenosis luminal.
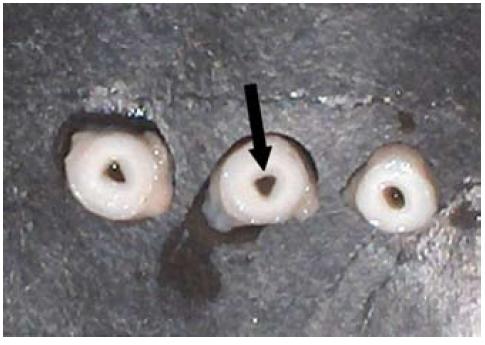
Figura 1b. Corte transversal de arterias carótidas. Nótese el engrosamiento de las paredes y la estenosis luminal.
Asimismo, se encontró congestión visceral, acentuada sobre todo, en pulmones e hígado. Otros hallazgos relevantes fueron marcada palidez en la mucosa gástrica e intestinal y lechos ungueales cianóticos.
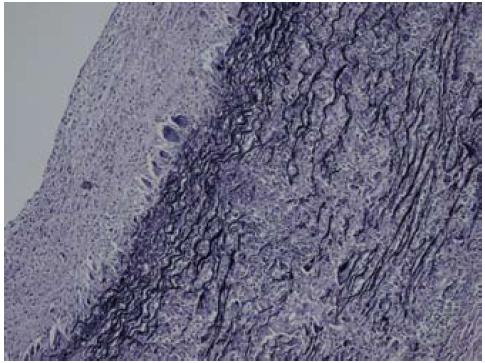
Figura 2a: Tinción para tejido elástico que muestra la fragmentación de dicha capa en la pared arterial.
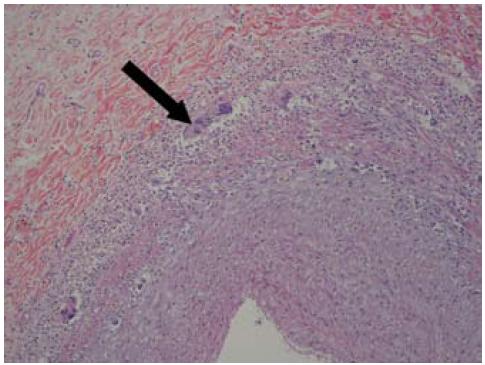
Figura 2b: Tinción hematoxilina eosina que muestra la infiltración de la pared y algunas células gigantes tipo Langhans (flecha).
En la reciente Conferencia Internacional de Consenso efectuada en Viena en 2005, se propuso una nueva clasificación para las vasculitis de la infancia.1 Esta toma en cuenta el tamaño de los vasos involucrados e incluye en el primer grupo las vasculitis que afectan predominantemente a los grandes vasos, donde la única entidad responsable es la arteritis de Takayasu.
La arteritis de Takayasu, conocida también como síndrome del cayado aórtico,2 enfermedad sin pulso o coartación inversa,3 fue descrita por primera vez en 1908 por el oftalmólogo japonés Makito Takayasu.4,5 Se trata de una vasculitis de grandes vasos1,6 que produce inflamación y estenosis de las arterias de mediano y grueso calibre, principalmente relacionada con el engrosamiento fibroso del arco aórtico con estrechamiento u obliteración virtual de las inserciones de los grandes vasos que emergen de este.
Arteritis de Takayasu en una niña / Vargas-Sanabria M y Valerio-Hernández M Algunos estudios señalan que las arterias más afectadas son las subclavias, hasta con un 93%,2 sin embargo, puede afectar las coronarias hasta en una frecuencia de un 10%. Su etiología es desconocida, aunque se ha intentado correlacionar sin éxito con marcadores como los antígenos de histocompatibilidad7 o factores infecciosos.8
El diagnóstico diferencial incluye otras aortitis inflamatorias (por sífilis, tuberculosis, lupus eritematoso sistémico, artritis reumatoide, espondiloartropatías, enfermedades de Buerger, Behçet, Cogan y Kawasaki), defectos estructurales (síndromes de Ehler Danlos y Marfan) y ciertas anormalidades aórticas (neurofibromatosis y fibrosis por radiación).7
De cualquier forma, desde el punto de vista histológico se puede aclarar con suficiente certeza: se trata de una panarteritis con infiltrado mononuclear adventicial con revestimiento perivascular de los vasa vasorum. Puede haber una inflamación mononuclear intensa de la media, acompañada en algunos casos por cambios granulomatosos, repleta de células gigantes de Langhans y necrosis central. Hay ruptura y degeneración de la lámina elástica. Sus cambios morfológicos pueden ser indistinguibles de los de la arteritis de células gigantes (de la arteria temporal),4 sin embargo, es mucho menos frecuente que ésta.
El perfil epidemiológico clásico de esta patología se ha centrado en individuos de origen asiático, de sexo femenino, cuyas manifestaciones iniciales de la enfermedad se manifiestan alrededor de los 25 años de vida.6-9,10 Su incidencia es de aproximadamente 2,6 casos por millón de habitantes por año, en poblaciones como la estadounidense.7 Por ello, la experiencia es casi anecdótica en territorios del hemisferio occidental.6,7 Sin embargo, también se ha dicho que no respeta región geográfica ni grupo étnico2; además de que el grupo etario afectado puede ser muy variable, pudiendo aparecer algunos pocos casos en lactantes.3
Las manifestaciones clínicas se correlacionan con el calibre de los vasos afectados y la forma más frecuente de presentación es la HTA,7,9-11 por estenosis de las arterias renales o por disfunción de los barorreceptores arteriales. Si bien la mortalidad es baja, y la sobrevida a 10 años elevada, la evolución clínica puede ser fulminante2 y cuando es así, la muerte generalmente sucede por ICC, IM, aneurisma roto, ECV o insuficiencia renal.7 Se ha dicho que la mortalidad pediátrica es elevada.3 El diagnóstico es eminentemente clínico, pues existen criterios como los de la Asociación Americana de Reumatología,6 pero el método diagnóstico más confiable es la angiografía,2,6 a pesar de que los hallazgos angiográficos varían de acuerdo con la región geográfica estudiada,8 dado que las biopsias arteriales son mucho más difíciles de obtener. La historia natural y el pronóstico de esta enfermedad están poco definidos.9
Como tratamiento médico se han utilizado los gluococorticoides, que si bien no se ha demostrado que mejoren la sobrevida, si inducen la remisión de la enfermedad por largos períodos. Asimismo, se han empleado agentes inmunosupresores como ciclofosfamida y metotrexate. Como tratamiento quirúrgico se han practicado utilizado los bypasses y las angioplastías, con un elevado porcentaje de reestenosis.6
En el paciente pediátrico la arteritis de Takayasu es la tercera vasculitis en frecuencia por detrás de la púrpura de Henoch-Schloein y la enfermedad de Kawasaki. La edad media de inicio es de 11,4 años, aproximadamente. El 75% de los casos corresponden a pacientes femeninas. La manifestación más frecuente es, al igual que en adultos, la hipertensión arterial, seguida de cerca por la cardiomegalia. Los vómitos también se han descrito como parte del cuadro clínico, pero con una frecuencia mucho menor.10 La mortalidad en este grupo alcanza el 33% y el tiempo de diagnóstico llega hasta 19 meses, mucho mayor que en adultos.6,7
Según la presentación de este caso, el abordaje inicial de la autopsia pudo haber girado en torno a la búsqueda de signos de intoxicación medicamentosa, debido al hecho de que la evolución se complicó inmediatamente después de administrar la prednisolona. Sin embargo, esto era poco probable, pues las reacciones adversas de los glucocorticoides suelen requerir mucho más tiempo para instaurarse,12 o bien el estudio post mortem pudo haberse centrado en la búsqueda de patologías con un cuadro clínico similar y fulminante, como la insuficiencia suprarrenal en su presentación de crisis2 mencionada en el expediente hospitalario. Pero después del examen macroscópico del cadáver, el hallazgo del engrosamiento y estenosis luminal de los grandes vasos fue determinante para orientar la indagación hacia un grupo de enfermedades muy características: las vasculitis. Además, los hallazgos sugerentes de congestión visceral e hipoperfusión tisular apuntaban a una ICC.
El caso descrito cumplió desde el inicio con las características macroscópicas de la enfermedad de Takayasu, pues la aorta y sus principales ramas estaban endurecidas12 y luego el diagnóstico fue ampliamente confirmado por los hallazgos histopatológicos de infiltración mononuclear, engrosamiento fibroso de la pared13 y engrosamiento de la íntima que obliteraba sobre todo las arterias coronarias; y zonas con células gigantes. Asimismo la congestión visceral generalizada, demostrada tanto micro como macroscópicamente hacía pensar en una ICC, que es la principal causa de muerte en la arteritis de Takayasu.12 Este caso resulta interesante, pues se trata de una paciente de 4 meses de edad, con una enfermedad cuya edad de presentación habitual es de 25 años. Era de raza blanca, para una entidad mucho más frecuente en los asiáticos. En algunos estudios la presentación en individuos caucásicos es tan baja como un 8 %. Por estas razones constituye una patología de muy difícil diagnóstico, sobre todo en el contexto demográfico de la paciente, pues algunas series señalan que el diagnóstico tarda mucho más en establecerse en pacientes pediátricos,7,8 por factores como la inespecificidad de los síntomas y la renuencia de los especialistas a efectuar estudios invasivos como la angiografía,8 primordial en la caracterización de esta enfermedad. Asimismo, la evolución generalmente es a largo plazo,8 sin embargo, en el presente caso, a escasos 4 días de haber presentado una sintomatología tan inespecífica como tos seca, la paciente sufre en el último día una descompensación súbita que la lleva a una muerte por insuficiencia cardiaca (Figura 3).
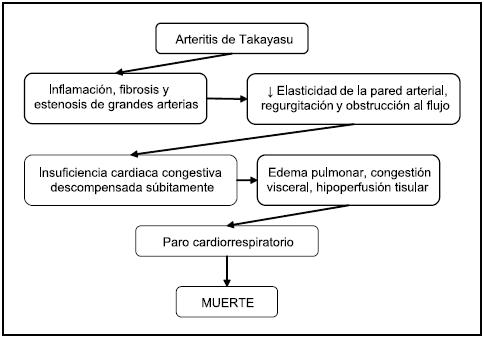
Figura 3: Diagnóstico fisiopatológico
En nuestro país, en Europa, Norteamérica, 13 Brasil9 y en general en el hemisferio occidental, un caso de arteritis de Takayasu en una lactante de 4 meses de edad, de raza blanca, y que evolucione de manera fulminante constituye un caso excepcional, pocas veces descrito en la bibliografía. El pediatra, al igual que el médico forense o anatomopatólogo, debe conocer esta entidad para tenerla en cuenta al abordar un caso con una presentación como la descrita y tener la capacidad de establecer el diagnóstico certera y rápidamente.
Abstract
We present the case of a previously healthy 4 monthsold child, , who suffered a 4 day illness, unspecific at the beginning, but during her last day showed signs of tissue hipoperfusion, had 2 cardicac arrests and died. The forensic autopsy and histological examination demonstrated great vessels vasculitis. This findings are compatible with Takayasu’s arteritis. Takayasu is a very uncommon disease in the occidental hemisphere, and is also unusual in caucasic children and uncommonly has a lethal outcome.
Referencias
1. Dillon MJ, Ozen S. A new international classification of childhood vasculitis. Pediatr Nephrol. 2006 Jul 4. [Epub ahead of print]. [ Links ]
2. Braunwald E, Fauci A, Kasper D, Hauser L, Longo D, Jameson J. Harrison: Principios de Medicina Interna. 15 ed. México: Mc Graw Hill, 2001. [ Links ]
3. Behrman R, Kliegman R, Jensen H. Nelson. Tratado de Pediatría. 16 ed. México: Mc Graw Hill Interamericana, 2001. [ Links ]
4. Cotran R, Kumar V, Collins C. Robbins: Patología estructural y funcional. 6 ed. México: Mc Graw Hill Interamericana. 1999. [ Links ]
5. Lazzarin P, Pasero G, Marson P, Cecchetto A, Zanchin G. Takayasu’s arteritis. A concise review and some observations on a putative case reported by Giovanni Battista Morgagni (1761). Reumatismo. 2005; 57:305-13. [ Links ]
6. Lacruz Pérez L. Granulomatosis de Wegener y arteritis de Takayasu. An Pediatr (Barc) 2005; 62:271-6. [ Links ]
7. Kerr GS, Hallahan CW, Giordano J, Leavitt RY, Fauci AS, Rottem M, Hoffman GS. Takayasu arteritis. Ann Intern Med. 1994; 120:919-29. [ Links ]
8. Sato EI, Hatta FS, Levy-Neto M, Fernandes S. Demographic, clinical, and angiographic data of patients with Takayasu arteritis in Brazil. Int J Cardiol. 1998; 66 Suppl 1:S67-70. [ Links ]
9. Mwipatayi BP, Jeffery PC, Beningfield SJ, Matley PJ, Naidoo NG, Kalla AA, Kahn D. Takayasu arteritis: clinical features and management: report of 272 cases. ANZ J Surg. 2005; 75:110-7. [ Links ]
10. Jain S, Kumari S, Ganguly NK, Sharma BK. Current status of Takayasu arteritis in India. Int J Cardiol. 1996; 54 Suppl 1: S95-100. [ Links ]
11. Sharma BK, Jain S, Radotra BD. An autopsy study of Takayasu arteritis in India. Int J Cardiol. 1998; 66 Suppl 1: S85-90. [ Links ]
12. Baumgartner D, Sailer-Hock M, Baumgartner C, Trieb T, Maurer H, Schirmer M, Zimmerhackl LB, Stein JI. Reduced aortic elastic properties in a child with Takayasu arteritis: case report and literature review. Eur J Pediatr. 2005;164:685-90. [ Links ]
13. Flórez, J. Farmacología Humana. 3 ed. Barcelona: Masson, 2001. [ Links ]

