










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.47 n.4 San José Oct. 2005
German F. Sáenz-Renauld
Resumen
Es notorio que en Costa Rica la frecuencia de las variantes más comunes de la hemoglobina (Hb) (S Y C) puede ser trazada desde África. Estas hemoglobinas son dos marcadores genéticos que han mantenido un equilibrado polimorfismo contra la presión selectiva de la malaria, en los países de origen o ancestrales. En la población caucásica son muy raras las mutantes estructurales de la Hb y se considera que nuestros indígenas no presentan ninguna alteración en elloci de la Hb. Un breve repaso antropológico, clínico, diagnóstico, genético y epidemiológico deja claro el verdadero problema de salud pública de estas enfermedades hereditarias de la hemoglobina, con especial énfasis en la drepanocitosis. Tangencialmente se destaca el problema de la deficiencia de la deshidrogenasa de la glucosa-6-fosfato en la etnia negra/negroide del país.
Descriptores: Hemopoyesis, genes de globina, hemoglobinas anormales, anemia hereditaria, síndromes drepanocíticos, haplotipos del gene drepanocítico.
Key words: Hemopoiesis, globin genes, abnormal hemoglobines, malaria, hereditary animia, sickle cell syndromes, sickle cell gene haplotipes.
Recibido: 5 de julio de 2005 Aceptado: 16 de agosto de 2005
Al igual de lo que acontece en la mayoría de los países de América, la distribución y frecuencia de las variantes estructurales de la hemoglobina (Hb) en Costa Rica, depende en gran medida de un factor étnico ligado al origen de sus poblaciones. Cuando la causa étnica se establece con claridad, como es el caso de las personas de raza negra, la distribución de los principales polimorfismos de la Hb en nuestro país, se pueden efectuar con relativa facilidad. Esto ha constatado en los varios estudios poblacionales, en donde también se ha determinado que las más frecuentes hemoglobinopatías son la Hb S y la Hb C, reflejo de la estructura antropológica de los diferentes grupos que forman nuestra población. La prevalencia que se observa en Costa Rica de las Hbs S y C indica, en parte, la influencia de algunas condiciones ecológicas en la tierra de origen, toda vez que estas dos Hbs son marcadores genéticos caracterizados por un equilibrado polimorfismo, en razón de la presión selectiva provocada por la malaria 1-3.
Si se hace una analogía con los hallazgos en el África centro-occidental, la cuenca del Caribe y otras regiones del América tropical con alto índice de ancestros africanos, constituye otra especie de "tierra de las hemoglobinopatías" 2, en la cual la Hb S asume la mayor importancia clínica y antropológica.
En América, la contribución africana ha sido predominante desde los inicios del siglo XVI, y cubre un período de más de 300 años, con base en la migración forzada de alrededor de 20 millones de esclavos. Por lo tanto, la presencia de hemoglobinas anormales es el producto de la inmigración de diversas poblaciones africanas, cuyos defectos hereditarios de la Hb son frecuentes 3. Esta realidad hace pensar que estos padecimientos se incrementarán aún más por las mezclas étnicas, las cuales en tiempos pasados estuvieron limitadas o controladas por la segregación racial. Sin embargo, al romperse esas cadenas sociales y asimilarse la etnodilución por las variadas y accesibles comunicaciones del tránsito humano, las circunstancias podrían variar ostensiblemente.
En la población caucásica de América son raras las hemoglobinopatías estructurales; por lo general, se trata de mutaciones privadas, esporádicas u ocasionales, y espontáneas ya con una relación genética distante 4. No existe polimorfismo de la Hb en la población autóctona, indígena o aborigen del país 5. Algunos hallazgos de ciertos marcadores genéticos permiten mantener vigente la hipótesis acerca del origen asiático-mongoloide del amerindio 1.
La colonización española de América, con la contribución de otras nacionalidades europeas, la inmigración compulsiva de africanos y la contribución del genoma aborigen, provocaron un entrecruzamiento reproductivo en diferentes extensiones, y originaron poblaciones heterogéneas híbridas, según el grado de flujo de genes 6-8, particularmente caucasoides o negroides hacia el genoma amerindio 9.
Diferencias en la composición étnica y en el grado de entrecruzamiento, y variaciones en la presión selectiva de la malaria de región a región, son las responsables de las diferentes frecuencias de las Hbs S y C en nuestros países. Al margen de la frecuencia de Hb S en la América Tropical (718%), en las islas y regiones que conforman la cuenca del Caribe, la prevalencia de la Hb C oscila entre el 1.2% y el 6.4%. En Costa Rica se aprecia una diferencia significativa al comparar la frecuencia de este marcador (Hb C) entre la población de raza negra de Limón (3.6%) y la negroide de Guanacaste (0.30%). Tal diversidad podría explicarse por movimientos migratorios, al suponerse que los negros que se asentaron en la costa norte del Pacífico debieron provenir de zonas del África en donde la prevalencia de la Hb C es relativamente baja.
Por otra parte, los inmigrante s jamaiquinos que llegaron contratados como trabajadores agrícolas a las costas del Caribe, eran descendientes africanos centro-occidentales: Ghana, Alto Volta (epicentro de la mutación), Liberia, Sierra Leona, Costa de Marfil, Togo, Benin y el oeste de Nigeria 2. Esta misma explicación podría aplicarse a otros países americanos donde se han encontrado frecuencias variables de Hb C entre la población de extracción africana.
Interacción de las Hbs anormales y la deshidrogenasa de la Glucosa-6-Fosfato (G6PD; Gd-): La deficiencia de la Gd y las Hbs anormales a menudo afecta a las mismas poblaciones expuestas a una presión selectiva por parte del Plasmodium falciparum. La existencia de ambos defectos, especialmente en raza negra, obliga a la determinación de la Gd como parte integral del diagnóstico de los desórdenes de la Hb 3,10. Los genes de estos dos trastornos se encuentran en diferentes cromosomas, siendo el de la Hb S autosómico codominante, en tanto que el de la Gd se halla ligado al sexo; su segregación, es independiente. La alta frecuencia del gene βS se mantiene en continentes como el africano, por la protección parcial del heterocigoto (Hb AS) contra los efectos del P jalciparum. De igual forma, la deficiencia de Gd (mutante Gd A- en África) se ha encontrado con alta frecuencia en áreas de malaria holoendémica, por lo que se conjetura que también desempeña un papel protector contra dicha enfermedad infecciosa 1,8. Las alteraciones metabólicas dadas por la deficiencia de Gd, y las fisicoquímicas, por la presencia de Hb S, serían las responsables de ofrecer un microambiente celular adverso al P. jalciparum 11; función similar se le ha conferido a la talasemia 12. Por otro lado, los investigadores jamaiquinos han referido que en la drepanocitosis la deficiencia concomitante de Gd no influye en la concentración de Hb, en el cómputo de reticulocitos, en la concentración de Hb F, en el cómputo de drepanocitos irreversibles, ni en la concentración de Hb plasmática, y no se ha encontrado tampoco relación entre la gravedad clínica y la existencia o ausencia de la deficiencia de Gd 13. Se considera que estos hallazgos en adultos deben ser necesariamente diferentes de lo que acontece en niños, entre quienes es más problemática la asociación Gd A-/Hb SS, por las frecuentes infecciones en esa etapa. La coexistencia del gene Gd A- posiblemente cause una ligera desventaja a los pacientes con Hb SS en Nigeria 14, y ciertamente, no mejora el curso de la enfermedad en el contexto genético y ambiental del África tropical. A esa misma conclusión se llega en Ghana, en donde se logra demostrar, bajo parámetros clínico-epidemiológicos, que la deficiencia de Gd en el paciente SS, lo hace más enfermo y le confiere mortalidad más temprana 15.
La situación en Costa Rica en torno a las Hbs anormales: La primera publicación sobre la existencia de Hbs anormales en Costa Rica apareció en la Revista Médica de Costa Rica de 1945, en donde Aguilar y Piedra 16 reportan el caso de una mujer de 26 años, soltera, mestiza, de la ciudad de Cartago, que ingresó en el Hospital San Juan de Dios por extrema esplenomegalia e ictericia. La comunicación de laboratorio indica abundantes eritrocito s alargados a manera de drepanocitos, reticulocitosis, eritroblastemia y una Hb de 8.8 g/dI. Los autores mencionan en esta enfermedad -no tanto anemia drepanocítica, sino doble heterocigota por Hb S y β talasemia-, que en 1941 Sáenz Herrera y Álvarez Iraeta, a la sazón médicos pediatras de aquel Hospital, ya habían observado por primera vez un caso de "drepanocitemia" en un niño puertorriqueño. En 1961, Elizondo 17 presentó ante el Congreso Médico Centroamericano, un caso de priapismo por drepanocitosis en un sujeto mestizo de padre panameño. Bicker et al. 18, en 1965, analizando en los Estados Unidos algunos especímenes sanguíneos de individuos costarricenses de raza negra, comunican un doble heterocigoto Hb C/G-Philadelphia y un caso de persistencia hereditaria de Hb F y, finalmente, Elizondo y Solano 19 en 1965, destacan un caso doble heterocigoto por Hbs S y C, en un individuo híbrido de Guanacaste, en el que se evidenció una coriorretinitis no tratada, alteraciones neurológicas y esplenomegalia. En fechas recientes se ha constatado el usual y moderado cuadro de esta enfermedad doble heterocigota, más deficiencia de GdA-, con una grave complicación ocular con retinitis proliferativa y hemorragia del vítreo 20.
La búsqueda intencionada de Hbs anormales se inició en Costa Rica en 1966, cuando Solano et al. 21 dan cuenta, en población híbrida del cantón de Santa Cruz, Guanacaste, de un 7.5% de muestras positivas por drepanocitos con la evidencia electroforética de un caso "homocigota" por Hb S. En 1967, Solano y Mainieri 22 señalan un 4.8% de Hb AS en la población de Liberia, Guanacaste. En 1968, Rivera y Sáenz 23, con la intención de obtener datos numéricos y estadísticos mínimos sobre la frecuencia de Hbs anormales en Costa Rica, encuentran un 1.4% de hemoglobinopatías en un total de 947 adultos de 13 poblaciones distribuidas en todo el territorio costarricense, destacándose en algunas una extracción caucásica, en otras híbrida, y en las menos, predominio de raza negra. En esta comunicación de los autores se menciona por vez primera la frecuencia de Hb C en Costa Rica (0.3%). Se destaca en este informe el uso de la electroforesis convencional a pH alcalino con tiras de acetato de celulosa, la aplicación de la prueba de solubilidad original para Hb S, la cuantificación de la Hb A2 por electroforesis en acetato y posterior elusión y, la dosificación de la Hb F por desnaturalización alcalina. Sáenz et al., en 1971 24, destacan en 621 individuos de raza negra, de uno y otro sexo, niños y adultos, un 8.2% de Hb S y un 2.4% para la Hb C. Esta información permitió, por primera vez, conocer con bastante exactitud la frecuencia de estos defectos en nuestra etnia negra.
Es importante destacar que Sáenz et al. 25, en el mismo año, habían demostrado la alta frecuencia (10%) de la deficiencia de la G6PD eritrocítica (Gd A-) en esa población. En 1973, Sáenz et al. 26 encuentran en Santa Cruz de Guanacaste un 8.9% de Hb S, 0.30% de Hb C. Este estudio realizado en 1702 muestras de escolares y estudiantes de secundaria, étnicamente de predominio mestizo, permitió observar dos casos de drepanocitosis, y un 2.6% de Hb A' 2, variante de cadenas delta, típicamente africanas.
Para 1980, con la creación del CIHATA (Centro de investigación en hemoglobinas anormales y trastornos afines, en la Universidad de Costa Rica), se llevó a cabo el más ambicioso proyecto de investigación en Costa Rica, en tomo a hemoglobinopatías y trastornos afines 27. Para ello se seleccionó estadísticamente un muestreo de 12.000 niños escolares, diferenciándoseles en forma empírica en caucásico-caucasoide, híbrido s o mestizos y de raza negra 28. En el cuadro 1 se indica la frecuencia de las Hbs S y C en dichas poblaciones, destacándose, una alta incidencia de esas Hbs en los escolares de raza negra o de extracción africana (mestizos, híbridos). No fue sino hasta en 1984 cuando fue posible detectar el primer caso homocigoto de Hb C, en un niño de raza negra que ingresó al hospital por esplenomegalia y sindrome ictérico 29.
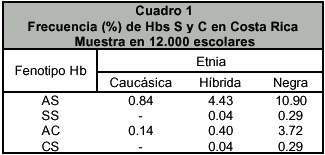
Hemoglobinopatías poco comunes en Costa Rica:
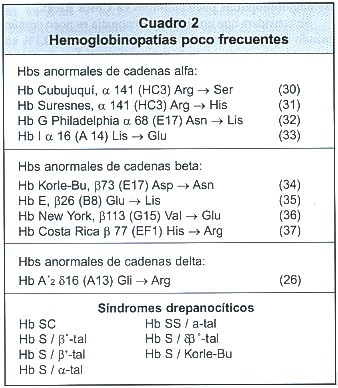
Se considera que una hemoglobinopatía es poco común cuando se observa con una frecuencia menor del 0.1 % en una población dada según la Oficina Mundial de la Salud, OMS 6,7. En atención a los resultados poblacionales pediátricos indicados 28, en Costa Rica la frecuencia en la población escolar general de la Hb S es del 2.5%; de la Hb C, del 0.4%; y de la variedad de beta talasemia, menor del 0.25%. Estas tres hemoglobinopatías son, por lo tanto, las más frecuentes y las que tienen mayor trascendencia clínica, sobre todo en sus formas homocigotas o doble heterocigotas, como serían los síndromes drepanocíticos tipo SS y SC, la interacción de la Hb S con genes de beta talasemia y la talasemia mayor. Hay hemoglobinopatías muy poco comunes y otras que aparecen como mutantes totalmente nuevas, siendo ambos hallazgos de gran interés como marcadores genéticos y antropológicos y, aún clínicos, para una determinada población.
Hasta el momento de esta publicación se han registrado en el país los defectos poco frecuentes que se enumeran en el cuadro 2.
Generalidades sobre la anemia drepanocítica (An):
En la drepanocitosis usualmente no se producen síntomas en los primeros seis meses de vida, en vista de que por ese tiempo la Hb predominante es la Hb F que es antipolimerizante y con fuerte afinidad por el oxígeno, al contrario de la Hb S. Luego de ese período el niño se presenta con palidez e ictericia, como resultado de la anemia hemolítica crónica asociada a la enfermedad. El agrandamiento del bazo y del hígado son características de la etapa infantil, como resultado de hemopoyesis extramedular o de la congestión secundaria por la sicklemia intravascular. El dolor (crisis vaso-oclusiva) es causado por el bloqueo de pequeños vasos por células densas y drepanocíticas. Se pueden presentar otras formas de crisis en esta enfermedad, como son la aplástica, la hiperhemolítica y la de secuestración esplénica. Una significativa complicación de la AD, especialmente en la edad infantil, es la predisposición a infecciones bacterianas con alto índice de mortalidad. En niños mayores puede haber retardo en el crecimiento y en el desarrollo de las características sexuales secundarias. Con el tiempo, cualquier órgano del cuerpo puede ser afectado por episodios recurrentes oclusivos, que son los cuadros clásicos de esta enfermedad.
Las células drepanocíticas y su papel oclusivo, se originan en la microcirculación por un mecanismo mucho más complicado de lo que se creía 38, con una adhesión inicial de reticulocitos y células densas al endotelio en las vénulas postcapilares. Luego se la adhieren leucocitos al endotelio con la formación de agregados heterocelulares. Todo ello origina hipoxia local y un incremento en la polimerización de la Hb S, con la propagación de la oclusión a la vasculatura adyacente. Ahora también se le da atención a la disregulación del tono vasomotor, por perturbaciones en mediadores vasodilatadores, tal como el óxido nítrico (NO). La deshidratación de los eritrocitos sicklémicos toma lugar por la activación de una o más de las vías de transporte de cationes. La homeostasis de cationes debe ser relevante en el tratamiento de la AD, ya que la inhibición de estos canales de transporte prevé la deshidratación de las células drepanocíticas, la formación de células densas y de los propios drepanocitos irreversibles, lo cual mejora los efectos del grado de hemólisis y de la adhesión endotelial. Estos y otros resultados se han podido destacar gracias a la experimentación con modelos de ratones sicklémicos transgénicos, en los cuales se ha logrado también la corrección de la AD con terapia génica 39.
La hemostasia se halla perturbada en las enfermedades drepanocíticas, muy en especial en la AD. Se ha sugerido que el endotelio activado es una de las fuentes de activación de la coagulación, con el apoyo a ese estado trombofilico de los eritrocito s fosfatidilserina positivos, que se consideran protrombóticos 40.
Algunos pacientes con AD presentan una enfermedad ligera o moderada y sobreviven a la edad adulta sin mayores complicaciones 38, 41-43. Los factores responsables para esta heterogeneidad clínica han sido objeto de muchos estudios. Se ha visto que tienen efecto favorable el incremento de la Hb F, otras mutaciones ligadas -por ejemplo- a sitios polimórficos en el cromosoma 11 que porta la mutación / βS (haplotipos), la coexistencia de alfa tal y factores ambientales. Particularmente, se ha constatado que pacientes con AD que presentan altos niveles de Hb F usualmente tienen un curso clínico moderado, al considerarse que la Hb F al no copolimerizar con la Hb S, inhibe en gran medida el fenómeno sicklémico 38,41,43. De acuerdo con la OMS 8, la drepanocitosis y la talasemia mayor son las enfermedades hereditarias de carácter letal más comunes en el mundo. Centenares de millones de personas son heterocigotos para estas dos principales hemoglobinopatías, y al menos nacen al año 200.000 homocigotos, letalmente afectados 6-8.
La frecuencia de entre un 20% y un 25% de heterocigotos para Hb S en ciertas regiones de África tropical, sugiere que aproximadamente el 4% de los matrimonios se encuentran en riesgo de engendrar un niño afectado y es probable que cerca de 100.000 niños drepanocíticos nazcan cada año en ese continente. En comparación, 10 hacen 1.500 anuales en los EEUU, 1.600 en el Caribe y 4.000 en Suramérica 8. Datos más recientes del Brasil muestran de nuevo la magnitud del problema de las hemoglobinopatías en los países con alta densidad de la etnia descendiente de África 44. Cada año, en ese país se reportan 2.600 nuevos casos de síndromes drepanocíticos mayores, 1.800 lo son por Hb SS, 400 por Hb SC y 400 corresponden al genotipo Hb S/β talasemia. Asimismo, se anotan 300 nuevos casos de β talasemia mayor. La AD, por lo tanto, constituye el tipo mαs frecuente de síndrome drepanocítico mayor; y para las autoridades de salud, este tipo de anemia debería ser una prioridad en los programas preventivos. La cifra del 7.5% de Hb AS, tasa frecuente en Costa Rica y en las poblaciones caribeñas, puede originar que de 170 uniones, una ocurra entre dos individuos AS, y que la frecuencia esperable de nacimientos con AD sea de 1:631. Mayores frecuencias podrían surgir al hacer los cálculos en poblaciones en las cuales la Hb AS ronde entre el 9% y el 11%.
La principal causa de muerte en la drepanocitosis la constituyen siempre los procesos infecciosos. En África, lo es por la malaria, y en el Caribe por la neumococcemia y otras septicemias bacterianas, a menudo asociadas al síndrome de secuestración esplénica 41. El mencionado estudio cohorte que se realizó en Jamaica 43 es el único en gran escala acerca de la historia de la drepanocitosis y los resultados de esta importante investigación, proveen un mejor cuadro de la historia natural de la drepanocitosis y de los resultados que se derivan al modificarse el medio ambiente.
Un mayor esfuerzo debería ponerse en la detección de heterocigotos y en el diagnóstico neonatal, con el propósito de permitir una identificación temprana y una protección al individuo portador. La detección del heterocigoto y un apropiado consejo genético y educativo para el paciente, así como un manejo adecuado del homocigoto, son aspectos que deberían estar integrados en el sistema primario de salud del país. La frecuente forma heterocigota de la Hb S (Hb AS) es asintomática, salvo excepciones derivadas por una baja substancial de la presión de oxígeno. Si se comparan parámetros fisiológicos, así como morbilidad y mortalidad, es posible concluir que el rasgo de Hb S (Hb AS) tiene un valor adaptativo neutro en ausencia de la presión selectiva de la malaria, por lo que el destino del gene beta S será el de desaparecer en aquellas regiones en donde la malaria ha sido erradicada.
Origen de la mutación de la Hb S (βS): Estudios antropológicos asociados a análisis biomoleculares, sugieren que el gene de la globina βS surgiσ por medio de un proceso de mutación de bases nitrogenadas que ocurrió entre 50 y 100 mil años, en los periodos paleolítico y mesolítico. Estos mismos estudios señalan África como el lugar probable en donde salió dicha mutación, coincidente con la presencia del horno sapiens sapiens (hace 40 mil años) y el neanderthalis (hace 100 mil años) 44. Se desconoce la causa que motivó ese cambio de una base nitrogenada (adenina) por otra (tímina), cuya traducción molecular provoca la sustitución del ácido glutámico por la valina en la posición número 6 de la globina β.
Un nuevo impulso para el estudio de la Hb S se logró con la introducción de técnicas de biología molecular, entre ellas, el uso de enzimas de restricción obtenidas de bacteriófagos específicos, lo que permitió admitir que la Hb S tiene múltiples orígenes. Estos análisis del gene βS han demostrado que al menos hay 5 tipos de Hb S, o sea, de drepanocitosis: SS-Benin, SS-Bantu, SS-Senegal, SSCamerún, SS-Árabe-indio, todas con la misma mutación, pero con diferente extensión en las lesiones moleculares ocurridas a lo largo del agrupamiento de los genes de globina del cromosoma 11 (5'-ε, yG, yA, ψβ, δ y βS- 3'). Estos descubrimientos esclarecieron en parte la diversidad clínica observada en distintos pacientes con AD, algunos con una evolución clínica benigna y otros con constantes complicaciones, y lograron establecer el origen multicéntrico del gene de la Hb S 44. (Cuadro 3).

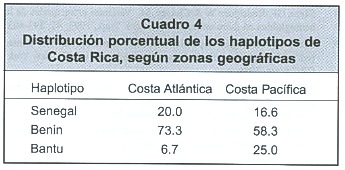
En América, los haplotipos del gene βS han demostrado ser de gran importancia como marcadores genιticos y antropológicos. Por ejemplo, en diferentes regiones de América Latina el estudio de los haplotipos βS ha permitido entender mejor las raíces ancestrales africanas de la población negra 44,47,48, Y ha contribuido también a aclarar las intrincadas aventuras del tráfico de esclavos desde el continente negro hasta el nuevo l. El polimorfismo genético de la βS ha sido establecido no solo entre las diferentes poblaciones negras de África y de América, sino también dentro de un mismo país, tal y como se demuestra en Brazil, Curazao, Senegal y Costa Rica, donde el haplotipo más frecuente de Hb S es el Benin (65%), seguido por el Bantu (20%) 48. Un reporte preliminar del CIHATA, facilitó valorar presuntivamente las características clínicas de la drepanocitosis en Costa Rica, al sugerirse que sus manifestaciones clínicas son graves en la zona de Guanacaste (litoral pacífico), donde la población negroide posee con mayor frecuencia los haplotipos Bantu y Benin perjudiciales, y que se observan menos complicaciones en pacientes de raza negra del litoral Atlántico (Limón), donde el haplotipo "inocuo" Senegal se encuentra en un 20% de la población 48. (Cuadro 4).
Los estudios sobre AD evolucionaron hacia otros objetivos, entre los que se destacan aquellos planteados para el uso de la quimioterapia, con el fin de estimular específicamente la síntesis de Hb F por los genes gama. La hidroxiurea es la que se ha usado con mayor éxito. El lector puede profundizar el tratamiento actual de la AD al consultar dos artículos recientes 38,45-47.
La evolución tecnológica de la biología molecular en el área de aislamiento y clonaje de genes de globina, se está dirigiendo a la realización de la terapia génica, al intentarse revertir el defecto desde las células hemotopoyéticas más primitivas. Esta tecnología ha permitido el análisis del ADN de la globina β para diagnósticos prenatales. Actualmente se está desarrollando con buen éxito el transplante de médula ósea no mieloablativa, mejor que con el TMO convencional (mieloablativo) 45.
Polimorfismo de la deshidrogenasa de la glucosa-6-fosfato en Costa Rica: Desde hace más de 25 años en Costa Rica se estudia el efecto hemolítico de la deficiencia de la deshidrogenasa de glucosa-6-fosfato (G-6-PD), especialmente en individuos de raza negra 49. En Costa Rica se detectó una frecuencia del 12.6% en el sexo masculino (hemicigotos), en 621 sujetos de raza negra 25. La frecuencia fue menor (4.3%) en una población híbrida o negroide de Guanacaste 26, y el estudio de 12.000 escolares de todo el país mostró globalmente que el 2.3% de los varones estaban afectados 28, sin importar de su origen étnico.
En 1982 se identificó un mutante de gran expresividad clínica que se designó Gd Puerto Limón 5°, y en 1983 se publicó otra variante patogénica, la Gd Santamaría 51.
Abstract
The frequecy of the commonest costarican Hgb varieties can the tracked to Africa. Hbg S and C, are genetic markers, that maintain a balanced polymorphism against malaria at the countries of origen. Among the caucasian population structural mutations of Hgb are unusual and it is felt that our indians exhibited no alteration at their Hgb loci.
The real public health problem of hederitary Hgb diseases, specially drepanocytosis, can be seen through a brief anthropological, clinical, diagnostic, genetic, and epidemiological review. The problem of G6PD deficiency in blacks and negroids is alluded to.
Referencias
1. Livinsgtone, FB. Anthropological aspects of the distribution of the human hemoglobin variant. En: Winter WP ed. Hemoglobin variants in human populations, vol 11. New York: CRC Press, 1986. [ Links ]
2. Cabannes, RJR., Fabritius, H., Sangare, A., & Kple-Faget, P. Hemoglobin variants: distribution in West Aftica. En: Winter WP ed. . Hemoglobin variants in human populations, vol 11. New York: CRC Press, 1986. [ Links ]
3. Colombo, B. & Martínez, G. Hemoglobinopathies; part 2 Tropical America. Clin. Haematol. 1981; 10: 730-756. [ Links ]
4. Sáenz GF, Rodríguez W & Chaves, M. Variantes estructurales de la hemoglobina en ibero américa. Rev. Biol. Trop., 1993; 41: 393-403. [ Links ] Sáenz GE Hemoglobinopathies in Central America. In W.P. Winter (ed.). Hemoglobin variants in human population, vol. 11. New York: CRC press, 1986.
6. Oficina Mundial de la Salud (OMS). 1972. Tratamiento de las hemoglobinopatías y de los trastornos afines. Informe de un grupo científico de la OMS. Serie Informes Técnicos No 509. [ Links ]
7. WHO. 1983. Community control of hereditary anaemias: memorandum from a WHO meeting. WHO Bull. 61: 63-80. [ Links ]
8. WHO. 1983. Report of the second annual meeting of the WHO Working Group on the community control of hereditary anaemias. Archbishop Makarios thalassaemia Centre, Cyprus 29-31 november. [ Links ]
9. Barrantes, R. Diversidad Genética y Mezcla Racial en los amerindios de Costa Rica y Panamá. Rev. Biol. Trop., 1993; 41: 379-384. [ Links ]
10. Sáenz, R., Jiménez, M., Chaves, M., Quintana, E & Sáenz GE Estudio sobre la coexistencia de Hemoglobina S y de la deficiencia de la Glucosa-6-PD deshidrogenasa en población de raza negra (Costa Rica). Rev. Cost. Ciencias. Med., 1986; 7: 305-310. [ Links ]
11. Motulsky, AG. Metabolic polimorfims and role of infectius diseases in human population. Hum. Biol. 1960; 32: 26-62. [ Links ]
12. Weatherall, D.J. Thalassaemia and malaria, revisseted. Aun. Trop. Med. Parasitol., 1997; 91: 885-890. [ Links ]
13. Gibbs, WN., Warde J. & Serjeant, GR. Glucose-6-phosphate dehydrogenase deficiency and homozigous sickle cell disease in Jamaica. Br. J. Haematol., 1980; 45: 73-79. [ Links ]
14. Bienzle, U., Sodeinde, U., Effong, EE & Luzzato, L. G6PD and sick le cell anemia: frecuency and features of the association in Aftican community. Blood, 1975; 46: 591-597. [ Links ]
15. Konotey-Ahulu, FID Glucose-6-phophate dehydrogenase deficiency and homozigous sickle cell anemia. N. Engl. J. Med., 1972; 287: 887-888. [ Links ]
16. Aguilar, E., Piedra, R.: "Un caso de drepanocitemia en una mujer costarricense". Rev. Med. Costa Rica, 1945; 139: 560-563. [ Links ]
17. Elizondo, J.: "Anemia de células falciformes. Reporte de un caso de priapismo por drepanocitosis (Costa Rica). Comunicación al IX Congreso Centroamericano de Medicina, 1961. [ Links ]
18. Biker, IN, Elizondo, J., Zomer, M.: "Hemoglobinopatías". Acta Med. Cost., 1965; 8: 3-6. [ Links ]
19. Elizondo, J.; Solano, L.: "Hemoglobinopatía S.C.". Acta Med. Cost., 1965; 8: 15-18. [ Links ]
20. Sáenz, GE, Ramírez, V & Chaves, M. Enfermedad por Hbs S y C y patología ocular. Acta Med. Cost., 1982; 3: 249-254. [ Links ]
21. Solano, L., Cabezas, M., Elizondo, L.: "Estudio sobre drepanocitosis y hemoglobina "S" en Santa Cruz de Guanacaste". Acta Med. Cost., 1966; 9: 59-62. [ Links ]
22. Solano, L., Mainieri, E: "Estudio sobre drepanocitosis y hemoglobina S en Liberia, Guanacaste". Acta Med. Cost., 1967; 10: 175-179. [ Links ]
23. Rivera, A., Sáenz, GF: "Datos numéricos y estadísticos mínimos sobre la incidencia de hemoglobinas anormales en Costa Rica". Rev. Med. Hosp. Nal. Niños, 1968; 2: 95-99. [ Links ]
24. Sáenz, G.E; Arroyo, G.; Jiménez, J.; Gutiérrez, A.; Barrenechea, M; Brilla, E, et al: "Investigación de hemoglobinas anormales en población de raza negra costarricense". Rev. Biol. Trop., 1971; 19: 251-255. [ Links ]
25. Sáenz, G.E, Brilla, E., Arroyo, G., Valenciano, E. & Jiménez, J.: "Deficiencias de la deshidrogenasa de la glucosa-6-fosfato (G-6PD) Eritrocitica en Costa Rica. 1. Generalidades sobre el defecto y hallazgos en población de raza negra". Rev. Med. Hosp. Nal. Niños, 1971; 6: 129-146. [ Links ]
26. Sáenz, GF, Alvarado, MA, Atrnetlla, E, Arroyo, G.; Jiménez, R & Valenciano, E.: "Investigación de hemoglobinas anormales en población costarricense del Guanacaste". Acta Med. Cost., 1973; 16:147-153. [ Links ]
27. Sáenz, GF, Elizondo, J., Arroyo, G., Jiménez, J., Montero, AG., & Valenciano, E. Diagnóstico de hemoglobinopatías y de trastornos afines. Enfoque población del problema. Bol. Of. Sanit. Panam., 1981; 90: 127-143. [ Links ]
28. Sáenz, G.E, Elizondo, J., Arroyo, G., Valenciano, E., Rojas, LE, Jiménez, J, et al. Hemoglobinopatías en 12.000 escolares costarricenses. Acta Med. Cost., 1980; 23: 89-99. [ Links ]
29. Sáenz RI., Sáenz GF., Muñoz, D., & Chaves, M. enfermedad homocigótica por Hb C. Primer reporte Nacional, 1984. Rev. Med. Hosp. Nal. Niños, 19: 25-30. [ Links ]
30. Sáenz, GF, Alvarado, MA, Elizondo, J, Arroyo, G, Atrnetlla, E, Jiménez, J et al. Chemical characterization of a new haemoglobin variant. Haemaglobin J-Cubujuquí (alpha2 141 (HC3) Arg-Ser). 1977. Acta Biochim. Biophys., 1977; 494: 48-50. [ Links ]
31. Sáenz, GF, Alvarado, MA., Arroyo, G, Montera AG., & Jiménez, J. 1978. Hemoglobin Suresnes in a Costa Rican Woman of SpanishIndian ancestry. Hemoglobin, 2: 383-385. [ Links ]
32. Sáenz, GF, Elizondo, J., Arroyo, G, Valenciano, E., Jiménez, J., Montera AG., et al. Hallazgo de la Hemoglobina G Philadelphia (alfa 68 (EI7) Asn-Lis) en Costa Rica: Consideraciones bioquimicogenéticas. Sangre, 1981; 26: 224-228. [ Links ]
33. Rodríguez, WE, Jiménez, J., Chaves, M., Montera, G, & Sáenz, GF: Hallazgo de la Hb I (? 16 (AI4) Lis-Glu) en Costa Rica. Rev. Cost. Ciencias Med., 1995; 16: 42-46. [ Links ]
34. Elizondo, J., Sáenz, GE, Alvarado, MA., & Ramón, M. Hallazgo de la hemoglobina Korle-Bu (alfa2 beta2 73 asp-asn) en Costa Rica. Sangre, 1976; 1: 54-56. [ Links ]
35. Sáenz, GF, Elizondo, J.,Colombo, B., Alvarado, MA., Arroyo, G, & Atmetlla F.. Hallazgo de la hemoglobina E (Beta 26 glu-lis) en Costa Rica. Sangre, 1977; 22: 652-655. [ Links ]
36. Sáenz, GF, Elizondo, J., Arroyo, G, Jiménez, J., & Montera, AG. Finding of the hemoglobin New York in Costa Rica. Hemoglobin, 1980; 4: 101-105. [ Links ]
37. Rodríguez, WE, Castillo, M., Chaves, M, Sáenz, GE, Gu, LH., Wilson, JB et al. Hemoglobin Costa Rica. The first example of a somatic cell mutation in a globin gene. Human Genet., 1996; 97: 829-833. [ Links ]
38. Stuart, MJ & Nagel, RL Sickle cell disease. Lancet, 2004; 364: 1343-1360. [ Links ]
39. Pawliuk, R., Westerman, KA & Fabry, ME. Correction of sickle cell disease in transgenic mouse models by gene therapies. Science, 2001; 294: 2368-2371. [ Links ]
40. Znaal, RF y Schrort, AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cell. Blood, 1997; 89: 11211132. [ Links ]
41. Serjeant, GR. Sickle cell disease. Oxford university press. 1985. [ Links ]
42. Serjeant, GR., Serjeant, BE., Forbes, M., Hayes, RJ., Higgs, DR. & Lehmann, H. Hemoglobin gene frequencies in the Jamaican population: a study in 100.000 Newboms. Brit. J. Haematol, 1986; 65: 253-262. [ Links ]
43. Serjeant, GR, Grandison, Y., Lowrie, Y., Mason, K., Phyllips, J; Serjeant BE, et al. The development of haematological chages in homozygous sickle cell disease: a cohor study from birth to 6 years. Br. J. Haematol. 1981; 48: 533-543. [ Links ]
44. Naoum, PC y Naoum, FA. Doenca das celulas falciformes. Savier, Sao Paulo, Brasil. 2004. [ Links ]
45. Valverde, K. drepanocitosis: terapia actual. Rev. Med. Hosp. Nal. Niños, 2004; 39:73-78. [ Links ]
46. Vichinsky, E. New therapies in sickle cell disease. Lancet, 2003; 360: 629-637. [ Links ]
47. Rodríguez, WE., Sáenz, GF., Josafovska, O., Nagel, RL. TheAfrican ports of origin of individuals of African descendent in Costa Rica: the use of? -gene cluster haplotypes. Page Rev. Invent. Clin. (Mexico), suplemento (Abstracts of XXV intemational Society of hematology congress CancÚll.) 1994. [ Links ]
48. Rodríguez, WE., Sáenz, GE, Chaves, M. haplotipos de al HbS: importancia, epidemiología y antropología clínica. Rev. Panam. Salud pública, 1998; 3: 1-8. [ Links ]
49. Sáenz, GE, Chaves, M., & Quintana, E. Polimomsmo de la deshidrogenasa de la glucosa-6- fosfato "G6PD" eritrocítica en Costa Rica. Sangre, 1988; 33: 261-264. [ Links ]
50. Elizondo, J., Sáenz, GF., Páez, CA, Ramón, M., García, M., Gutiérrez, A., et al. G6PD- Puerto Limón: a new deficient varíant of glucose-6-phosphate dehidrogenasa associated with congenital nonspheracytic hemolytic anemia. Human Genetics, 1982; 62: 110-112. [ Links ]
51. Sáenz, GF., Chaves, M., Barrantes, A., Elizondo, J., Montera, AG & Yoshida, A. A glucosa-6-phosphate varíant, Gd(-) Santamaría in Costa Rica. Acta Haemat., 1984; 72: 37-40. [ Links ]
Exdirector del CIHATA-UCR. Hospital San Juan de Dios.
Abreviaturas: Hb, hemoglobina; Hb SS, drepanocitosis; AD, anemia drepanocítica; Hbs S y C. variantes estructurales de la hemoglobina; Gd-. mutante africana de la G6PD (deshidrogenasa de la glucosa-6-fosfato); CIHATA, Centro de Investigación en Hematología y Trastornos Afines.
Correspondencia: Dr. German F. Sáenz Renauld. MQC, MSci. Apartado 5980-1000. Correo electrónico: labsare2@racsa.co.cr

