










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Médica Costarricense
On-line version ISSN 0001-6002Print version ISSN 0001-6012
Acta méd. costarric vol.47 n.1 San José Jan. 2005
El enfoque del paciente ictérico
Luis D. Quesada, Henry Zamora,Alfredo Martén (QdDG)
Resumen
Se presenta una revisión actualizada del metabolismo de la bilirrubina, las causas de la hiperbilirrubenemia, la clasificación implícita de las mismas y el abordaje del paciente con enfermedad hepatobiliar e ictericia según se maneja en nuestro medio en forma óptima de acuerdo a los recursos disponibles.
Descriptores: ictericia, hepatopatía, enfermedad biliar.
Recibido: 26 octubre de 2004 Aceptado: 30 de noviembre de 2004
La ictericia se define como la coloración amarillenta de la piel, las escleras y las mucosas, como consecuencia de una elevación anormal en la concentración sérica de bilirrubina (Br) 1-3.
En términos generales, los individuos con cualquier tipo de daño hepático, se encuentran ictéricos en algún momento de la evolución de dicho daño. Ciertamente, la mayoría de las veces puede considerarse ictericia y hepatopatía como sinónimos, como de hecho sucede con frecuencia. Sin embargo, es importante recordar, que algunos pacientes con enfermedad hepatobiliar no presentan ictericia, y asimismo, algunos pacientes con ictericia, no presentan enfermedad hepatobiliar 2,3.

La ictericia es una entidad clínica, conocida por lo menos desde los tiempos de Hipócrates, como consta en sus tratados 1. Para la época de William Osler, se podía distinguir con cierta veracidad, entre las causas obstructivas del árbol biliar y las causas no obstructivas de la ictericia1. Sin embargo, fue poco después de la mitad del siglo anterior, que se pudo definir con mayor precisión el metabolismo de la bilirrubina, se perfeccionaron las técnicas de análisis bioquímico, y se diseñaron métodos de diagnóstico por imágenes, que aclararan la etiología de la ictericia en la mayor parte de los pacientes 3, 4.
Existen varias maneras válidas de abordar a un paciente ictérico. El objetivo del presente artículo, es plantear un enfoque diagnóstico del enfermo con ictericia, según se hace de forma convencional y protocolaria en nuestro centro. La misma se basa en el concepto general de las enfermedades hepatobiliares, en el conocimiento propio adquirido durante el tiempo, en la disponibilidad de recursos diagnósticos existentes y finalmente, en el análisis concienzudo e integración de la literatura científica aceptada, respecto del tema.
Metabolismo de la bilirrubina
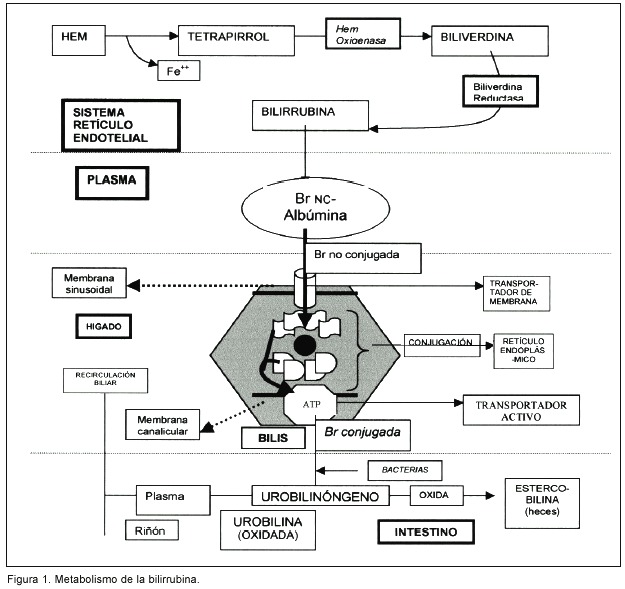
La bilirrubina es un pigmento tetrapirrólico proveniente del metabolismo del grupo "hem" de la hemoglobina (Hb) y otras hemoproteínas no hemoglobínicas. 2, 5 El hem está constituido por protoporfirina IX (unión de cuatro pirroles) y una molécula de hierro (Fe ++) 1, 3. Al liberarse el hem de la respectiva proteína, el hierro es reciclado y el anillo tetrapirrólico se abre de forma lineal, para ser convertido en biliverdina (por medio de la enzima hem oxigenasa). Ésta es rápidamente transformada en bilirrubina, por medio de la biliverdina reductasa. Todo esto sucede en el sistema retículo endotelial extrahepático, principalmente el bazo y la médula ósea 2,5,6. Un 70-80% de la Br proviene de la Hb de los eritrocitos senescentes, mientras que un 20-30% de la misma tiene su origen en proteínas diferentes a la Hb, que contienen grupo hem (principalmente en hígado: citocromo oxidasas y catalasas) 2,6. En condiciones normales, la contribución de la mioglobina a la formación de Br es casi nula.
La Br es una sustancia lipofílica y potencialmente tóxica. Requiere de un proceso de conjugación a formas hidrosolubles para ser eliminada. Se encuentra en el plasma en equilibrio entre la forma libre y la unida estrechamente a la albúmina 2,5. De esta última forma llega al sinusoide hepático por medio de la doble circulación hepática (arterial y portal) se libera de la albúmina e ingresa al hepatocito (por su membrana basolateral) por medio de un transportador de membrana específico y posteriormente es transportada al retículo endoplásmico por los ligandos (proteínas ligadoras específicas de la Br y de algunas otras sustancias) 2,3,5. En el retículo endoplásmico, es conjugada con uridina difosfato (UDP) y ácido glucurónico por medio de una enzima fundamental en el proceso llamada UDP- glucuronil transferasa, para formar un 80% de diglucurónidos de Br y un 20% de monoglucurónidos de Br. Menos del 1% de la Br en la bilis se encuentra en forma no conjugada 5,6. Luego, la Br conjugada (hidrosoluble) viaja hasta la membrana apical y a través de un transportador activo iónico (bomba ATPasa), es secretada al canalículo biliar para mezclarse con el resto de las sustancias que constituirán la bilis 5-7.
Una vez en el intestino, la Br conjugada es desconjugada por la flora bacteriana en el íleon terminal y en el colon, para formar pigmentos incoloros (urobilinógenos) 2,3. El 80% de los urobilinógenos, siguen su trayecto intestinal y se denominan estercobilinógenos –que también son incoloros. Una parte de estos últimos se oxidan para formar el pigmento que le da el color típico a las heces: estercobilina. El 20% de los urobilinógenos se reabsorbe al plasma, donde una fracción vuelve al hígado y recircula en la bilis; la otra se excreta en orina, donde una parte, también es oxidada a urobilinas 1,5. (Figura 1)
Determinación del nivel de bilirrubina
Normalmente la concentración de Br sérica no supera 1.5 mg/dl (con <5% de Br conjugada) 1,6. Se hace clínicamente manifiesta cuando el nivel alcanza los 3 mg/dl y muchas veces es el primer signo de hepatopatía.
El método más comúnmente usado en la práctica clínica para medir el nivel de Br, es el de la reacción diazo o método de van der Bergh 1,2,8. El mismo consiste en dos etapas, ambas involucran al ácido sulfanílico diazoado (diazo:doble enlace de sulfuro) 1,2,8.
La Br total presente en el suero, reacciona con el ácido sulfanílico para producir una molécula de azodipirrol, coloreada, que es medible por medio de espectofotometría 1,2. Sin embargo, la Br conjugada reacciona de forma más rápida (al cabo de un minuto), en medio acuoso. A esto se le llama reacción directa y de ahí el nombre de fracción directa de la Br. Por otro lado, la Br no conjugada reacciona de forma más lenta con el ácido sulfanílico. Esta reacción se puede acelerar al adicionar un agente catalizador al suero, como urea o etanol. Esto hace que la Br total reaccione para formar el azodipirrol cromogénico en aproximadamente treinta minutos. Para entonces determinar la fracción de Br indirecta, se resta la fracción de Br "directa" (reacción rápida en medio acuoso), del valor de Br "total" (reacción con agente acelerador) 2,3,8.
Es necesario aclarar que la Br "directa" no corresponde con absoluta exactitud, a la fracción sérica conjugada, ni la Br "indirecta" a la fracción no conjugada 1,3. Existen métodos más precisos, como la cromatografía líquida, pero que son más complejos, más caros y menos disponibles 2,3,8. Por tanto, el método de van der Bergh es universalmente aceptado para diferenciar las fracciones séricas de la Br.
Causas de hiperbilirrubinemia
I. Hiperbilirrubinemia de predominio indirecto
A. Sobreproducción
Hay varias entidades que pueden producir un aumento de la producción de Br. En general, corresponden a trastornos de enfoque hematológico, a saber: la hemólisis, la eritropoyesis ineficaz, la reabsorción de grandes hematomas y las transfusiones masivas 1,9. Rara vez sobrepasan los 5 mg/dl de Br sérica total 2,8. En todos estos pacientes, las pruebas bioquímicas hepáticas, suelen estar dentro de límites normales y la fracción de Br que se eleva de manera predominante es la indirecta.
B. Captación defectuosa
Aunque infrecuente, la captación defectuosa de Br es una causa de hiperbilirrubinemia de predominio indirecto. El agente más comúnmente relacionado con la inhibición competitiva de Br a nivel del transportador de membrana del hepatocito, es la rifampicina 1. El síndrome de Gilbert (ver adelante), también tiene un componente menor de captación defectuosa a nivel de la membrana del hepatocito 6,8.
C. Conjugación disminuida
Las enfermedades que cursan con una disminución en la conjugación de Br 4,6,8 se pueden agrupar en:
1. Ictericias del recién nacidoa. Cligler-Najjar I
b. Cligler-Najjar II
2. Ictericias familiares no hemolíticas
a. Síndrome de Gilbert
En todas estas entidades, el defecto genéticamente determinado, está en la actividad de la UGT-1 (UDP-glucuronil transferasa), pero lo que varía es el grado de disminución de dicha actividad 2,4,8. En el cuadro 1 se muestran las características más importantes de las tres entidades 2,3,6.
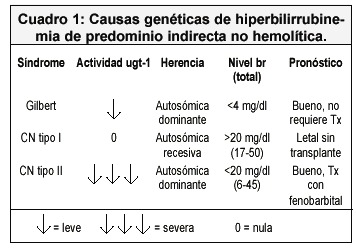
El síndrome de Gilbert es más frecuente en adultos, y se presenta en aproximadamente en el 10-12% de la población blanca 1,6. Es un síndrome y no una enfermedad.
El estrés físico o emocional, la deshidratación y el ayuno suelen evidenciar el trastorno al elevar la cifra de Br sérica y hacer la ictericia visible. La "prueba de ayuno" sirve para confirmar el diagnóstico de síndrome de Gilbert 2,3,8. El paciente es sometido a una dieta de no más de 100 g de carbohidratos por día por 48 horas. Si la cifra de Br se duplica en ese lapso la prueba es positiva 1,8. Con frecuencia se detecta de forma incidental, en pruebas de laboratorio realizadas por otra razón.
Ii. Hiperbilirrubinemia de predominio directo
Dentro de las entidades con aumento principal de la fracción directa de la Br (o para efectos prácticos, conjugada), virtualmente todas son colestasis4 en su comportamiento fisiopatológico, aún cuando el daño sea predominantemente hepatocelular, o no exista obstrucción mecánica. Sin embargo, para ordenar el aborde y manejo, se pueden clasificar como se muestra en el cuadro 2.

A) Trastornos hereditarios de la excreción de Br
Tanto en el síndrome de Dubin-Johnson como en el de Rotor, aumenta el nivel de Br sérica a < 7 mg/dl, con un franco predominio de la fracción directa 1,2,8. Ambas entidades se deben a un trastorno en el almacenamiento y/o la excreción de la Br conjugada. Ambas son poco frecuentes y de herencia autosómica recesiva. Se diferencian por el hecho de que en el Dubin-Johnson se acumula melanina (un pigmento oscuro) en los hepatocitos, lo cual le da al hígado un color negruzco. Ambas tienen función hepática normal y un buen pronóstico. No requieren tratamiento 1,3.
B) Colestasis intrahepática
Son múltiples las enfermedades hepáticas que cursan con colestasis intrahepática 1-4,8 y va más allá del propósito de esta monografía, detallar a fondo cada una de ellas. Sin embargo, es útil ubicarlas en grandes grupos. (Cuadro 3).

C) Colestasis extrahepática ( siempre con obstrucción mecánica al paso de la bilis)
En este grupo se encuentran las enfermedades que obstruyen el flujo de la bilis, en las vías biliares extrahepáticas, sea intrínseca o extrínsecamente 1-4,8. (Cuadro 4)

Abordaje diagnóstico
Anamnesis y examen físico
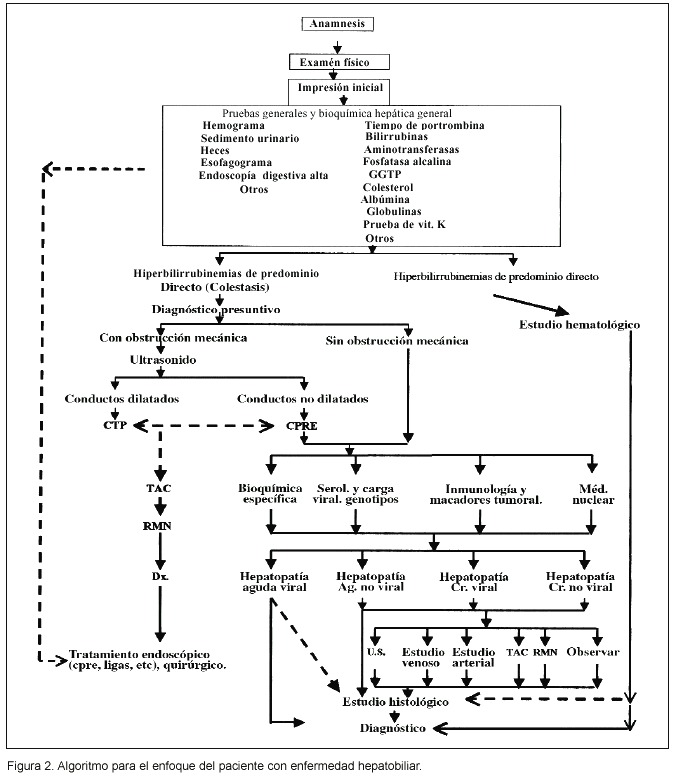
Tal y como se resume más adelante en la figura 2, la historia clínica, es el primer paso en el abordaje de un paciente ictérico y quizás el más importante, pues esto orientará sobre el cuadro en general y dictará los exámenes de laboratorio y gabinete pertinentes por escoger 4,6 . Es aquí donde deberá recopilarse toda la información clínica pertinente. Deberá investigarse lo siguiente:
2. Síntomas: dolor abdominal; fiebre; tiempo de evolución de la ictericia; pérdida de peso; coluria y acolia
3. Signos: datos de hepatopatía crónica; exploración abdominal (dolor, masas, líquido); estado del sensorio (encefalopatía); cicatrices abdominales; Ascitis.
Pruebas de laboratorio
El siguiente objetivo, luego de una anamnesis y un examen físico adecuados, es determinar la fracción predominante de Br 2,4,8 . Además es importante solicitar en este momento, pruebas bioquímicas hepáticas específicas y un hemograma 4.
La fosfatasa alcalina (FA) y la gamaglutamil transpeptidasa (GGT), son marcadores de excreción biliar y suelen estar aumentados de forma significativa en los procesos colestásicos en general. Aunque la FA puede elevarse por otras causas no relacionadas con hepatopatías, la GGT es casi exclusiva del hígado y confirma el origen hepático o no de la FA 1,8,10.
Las aminotransferasas o transaminasas (alanino aminotransferasa o ALT y la aspartato aminotransferasa o AST) son enzimas hepatocelulares que se elevan en casi todos los trastornos de necrosis hepatocelular de forma temprana (es generalmente el primer marcador de alteración hepatobiliar). Sin embargo, su aumento refleja daño celular directo (inflamación o necrosis). La AST es tanto citosólica como mitocondrial y se encuentra además en el músculo esquelético y en el miocardio. La ALT que es exclusivamente citosólica, es prácticamente específica del hígado 2,8,10.
Las proteínas séricas (especialmente la albúmina), y las pruebas de coagulación (especialmente el tiempo de protrombina o TP), se han catalogado tradicionalmente como pruebas de síntesis hepática 1,2,8.
La albúmina es sintetizada en el hígado, pero su disminución en plasma puede deberse a muchos factores extrahepáticos, principalmente pérdidas renales y poca ingesta proteica 1,8,11. Al tener una vida media de aproximadamente 20 días, es útil para valorar severidad y pronóstico de enfermedades hepáticas crónicas 2,3,8. Las globulinas suelen estar aumentadas en la enfermedad hepática crónica y el patrón de elevación puede sugerir la etiología: hepatitis por autoinmunidad inmunoglobulina G, cirrosis biliar primaria Inmunoglobulina M, enfermedad hepática alcohólica Inmunoglobulina A 2,8.
El TP es dependiente de la actividad de los factores vitamina K dependientes (I, II, V, VII, X), y es un marcador muy sensible en las enfermedades hepáticas agudas y de valor pronóstico significativo 1,2,8,10 . Puede verse aumentado en las enfermedades que afectan la capacidad de síntesis del hígado o en la deficiencia de vitamina K. La prueba de vitamina K es útil para diferenciar entre ambas etiologías: la administración parenteral de vitamina K mejora el TP en las ictericias por obstrucción mecánica con deficiencia de la misma, por mala reabsorción a nivel de la circulación entero hepática; no así cuando la función de síntesis del hígado está alterada 3,4.
El hemograma puede ser de utilidad, para determinar si el origen de la ictericia es hematológico (hemólisis) 1,2,4. Puede también poner en evidencia un proceso infeccioso agudo (leucocitosis o leucopenia), o entre otras más, sugerir hiperesplenismo (bi o pancitopenia) 1,4.
Serología, carga viral y genotipage
La serología viral puede aclarar la etiología de una enfermedad hepática aguda o crónica y es vital para el algoritmo diagnóstico, tal y como se presenta en este modelo de trabajo 4,12. Actualmente en nuestro medio se dispone de serología, para los virus hepatotróficos productores de hepatitis A, B, C, D, E, CMV y mononucleosis infecciosa. En el caso del virus de la hepatitis C, son de particular interés las determinaciones de carga viral, y la determinación del genotipo viral.
Pruebas inmunológicas y marcadores tumorales
Al igual que la serología viral, la carga viral de algunos virus, su genotipo, y los marcadores específicos de tipo inmunológico, pueden orientar respecto del origen y la severidad de una enfermedad hepatobiliar determinada. Se utilizan el factor antinúcleo y los anticuerpos antimúsculo liso para sustentar una hepatopatía por autoinmunidad y los anticuerpos anti- mitocondria para confirmar una cirrosis biliar primaria 4,8. También complementan la inmunoelectroforesis de proteínas, el complemento sérico, el anti ADN de doble cadena, el factor reumatoide, el antígeno hígado riñón, la alfa fetoproteína, el antígeno carcino embriónico, el CA 19-9 y el CA 125.
Otras pruebas
Existen otras pruebas mucho más específicas para enfermedades determinadas. Por ejemplo la céruloplasmina, el cobre sérico y la cupruria de 24 horas para la enfermedad de Wilson 1,2,10; los índices férricos y la transferrina para la hemocromatosis 3,10 y el nivel de alfa-1-globulina para la deficiencia de dicha enzima 1-3,10. También existe la posibilidad de hacer genotipo para la deficiencia de alfa-1- antitripsina.
Estudios de imágenes (directos e indirectos)
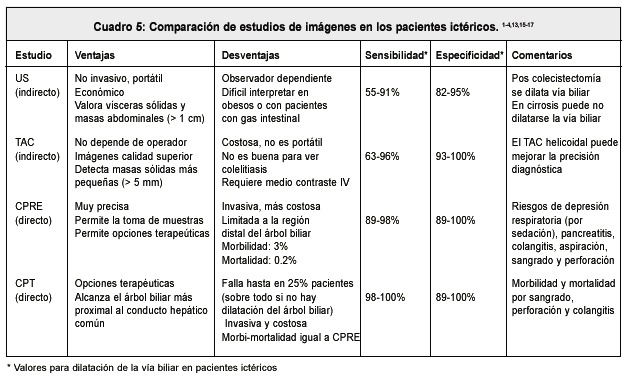
En los últimos 20 años, los estudios radiológicos y de imágenes, se han desarrollado de forma extraordinaria. Existen varios estudios de imágenes indirectas (tomografía axial computadorizada, ecografía) y directas (colangiopan-creatografía retrógrada endoscópica, CPRE y colangiografía transhepática percutánea, CTP), cuya utilidad en pacientes ictéricos es la de poder valorar la integridad o no de la vía bilio pancreática, determinar la presencia de lesiones espacio ocupantes intra y extra hepáticas, pancreáticas y ofrecer la posibilidad de realizar variados procedimientos terapéuticos 1,10,13. Todos estos procedimientos, tienen ventajas y desventajas. En el Cuadro 5 se presenta una comparación entre las técnicas de imagen usadas en nuestro medio. La disponibilidad de los métodos, y la experiencia personal en cada centro, en mucho determinada por la capacidad dependiente de operador, definirá, en la mayoría de los casos, el protocolo por seguir, definiéndose así cuál o cuáles estudios de imágenes y en qué secuencia, se van a llevar a cabo 13. Cuadro 5
Estudios endoscópicos de tamisaje
El estudio de endoscopía alta de choque (esófago gastro duodenoscopía), no es el más útil en la determinación etiológica de la ictericia, pero puede evidenciar datos de hipertensión portal (várices gastro-esofágicas y/o gastropatía de la hipertensión portal), que sugieren fuertemente el diagnóstico de una hepatopatía crónica 1-31.
Biopsia hepática
La biopsia hepática es el estándar de oro diagnóstico en prácticamente todas las hepatopatías (con algunas excepciones en las que no es necesaria) 14. Revela información sobre la arquitectura lobulillar y celular, la carga de hierro y cobre, la integridad de los conductillos biliares y la presencia de fibrosis, inflamación, infiltración y mitosis, entre muchos otros aspectos 1,2,14 . Tiene una tasa baja de complicaciones, morbilidad < 0.5% (sangrado, perforación, biliperitoneo) y mortalidad < 0.1% 1-3,13,14.
Flujograma de trabajo para las ictericias
Desde un punto de vista académico, en la figura 2, ofrecemos un esquema de nuestra visión actual, respecto de la secuencia que se sigue en nuestro centro para estudiar un paciente con probable enfermedad hepato pancreato biliar, en principio ictérico. No obstante, en bastantes casos, estos mismos pacientes pueden presentarse sin ictericia. No desconocemos que lo propuesto puede seguirse fielmente, o en muchos casos, simplificarse con cortocircuitos. Éstos pueden acortar el tiempo, para llegar a un diagnóstico correcto y, por lo tanto, la toma de decisiones. Esto sucede muy frecuentemente en los Servicios de Urgencias, de manera válida, ante las características de los pacientes y sus enfermedades que ahí se manejan. Esto será objeto de otra comunicación. Figura 2
Idealmente, el diagnóstico se orienta con la clínica (anamnesis y examen físico), como ya se ha enfatizado antes. Se debe solicitar el nivel sérico de Br total y fraccionada, y así poder definir la fracción que predomina. Para estos efectos, el paso crítico en el metabolismo de la Br, es su excreción biliar. Ya sea de manera proximal, en su meta por salir del hepatocito y verterse al canalículo biliar, o de manera distal, cuando encuentra cualquier obstáculo, en su viaje desde el canalículo, hasta su llegada a la segunda porción del duodeno, a través de la ampolla de Vater. Para definir el predominio de la fracción indirecta debe existir al menos 80% de ésa y en el caso de la directa, debe existir al menos 50% de esta última 18. Además, concomitantemente, se pide un hemograma y las pruebas bioquímicas hepáticas.
Si la fracción de bilirrubina predominante es indirecta y la hemoglobina está baja, el origen de la ictericia es probablemente hematológico y los estudios se orientarán en ese sentido. Si por el contrario la hemoglobina es normal, se debe pensar en un síndrome de Gilbert y corroborar el mismo con una prueba de ayuno 1,8.
Si la fracción de Br directa es la predominante (o la hiperbilirrubinemia es mixta), el trastorno es colestásico. En este caso, y tomando en cuenta toda la información clínica y de laboratorio que se tenga en ese momento, se puede suponer si se trata de una colestasis con obstrucción mecánica al paso de la bilis o si por el contrario se trata de una colestasis bioquímica o funcional, sin obstrucción mecánica.
Si al hacer el diagnóstico presuntivo la alternativa más probable pareció una colestasis con obstrucción mecánica al paso de la bilis, se impone realizar un ultrasonido (ecografía) 8,15,16, que nos puede aclarar si la vía biliar intra extrahepática está dilatada, e incluso revelar la causa de la obstrucción. Si la vía biliar esta dilatada, se recomienda realizar una CPRE o una CTP 8,17 para aclarar la etiología de la obstrucción (si no se ha aclarado aún), y para ofrecer opciones diagnósticas (biopsia, cepillado, aspirado de la vía biliar), y terapéuticas específicas (papilotomía, barrido con extracción de cálculos, colocación de férulas intraductales) 1,17. Si estos estudios son negativos, entonces es pertinente realizar una biopsia hepática 1,8,14. Si la vía biliar no está dilatada y existe una alta sospecha de obstrucción mecánica, se deben solicitar anticuerpos antimitocondriales para descartar una cirrosis biliar primaria 8,10. Si aún este marcador es negativo, se procederá a realizar en este momento la CPRE o CPT 4,8. De ser también normal el estudio elegido, se procederá a manejar como una colestasis sin obstrucción mecánica al paso de la bilis.
Por otro lado, si la sospecha clínica es que se trata de una colestasis funcional, sin obstrucción mecánica, se impone decidir si se trata de una hepatopatía aguda o crónica, y en cada caso, si la etiología es o no viral 1,3,4. Consideramos que ubicar a los pacientes que nos ocupan en alguna de estas cuatro categorías significa haber avanzado enormemente en la conceptualización de su enfermedad: hepatopatía guda No Viral, hepatopatía aguda viral, hepatopatía crónica no viral y hepatopatía crónica viral. Cinco grupos de pruebas y una actitud voluntaria particular ayudan a ello:
1. La serología, la carga viral y el genotipage, según correspondan (A, B, C, D, E, GCMV, Epstein-Barr)
2. Las pruebas inmunológicas y los marcadores tumorales, según falten y sean necesarios en este momento (anti mitocondria, anti músculo liso, inmunoelectroforesis de proteínas, complemento sérico, anti ADN de doble cadena, anti núcleo, factor reumatoide, mono test, antígeno hígado-riñón, y otros; alfa fetoproteína, antígeno carcino embriónico, CA 19-9, CA 125 )
3. Las pruebas específicas (céruloplasmina, cupruria, índices férricos, α-1 globulina, pruebas para enfermedades de depósito, etc.)
4. Los estudios de imágenes (ultrasonido, TAC y resonancia magnética nuclear)
5. La esófago gastro duodenoscopía.
6. Observación.
Si se trata de una hepatitis viral aguda, generalmente no es necesario realizar una biopsia hepática al paciente 1 , pues muchas veces puede mostrar un patrón histológico complejo, para el patólogo no especializado en el campo. Sin embargo, habrá casos en que sea necesario practicarla 14 (estudios inmunohistoquímicas, tinciones especiales, etc.). Las otras tres alternativas sí deben corroborarse con biopsia hepática, siempre y cuando las pruebas de coagulación y el estado hemodinámico del enfermo lo permitan, incluyendo las hepatitis agudas no virales (por ejemplo, las tóxicas y por fármacos) 4,8,14. En la mayoría de las hepatopatías crónicas, se requerirá corroboración e integración de la información clínica, de laboratorio y de gabinete, antes de realizar la biopsia hepática.
Con toda esa información, más la biopsia, la posibilidad de un diagnóstico correcto, y de la escogencia de un tratamiento médico, quirúrgico o combinado adecuado, es muy probable. Como se mencionó al principio, éste es un planteamiento arbitrario; no es, por mucho, la única ruta para llegar a la meta. Sin embargo, esperamos que sea de utilidad para todos los colegas y compañeros, el conocer nuestra manera de enfocar y abordar el curso diagnóstico y algunas medidas terapéuticas iniciales, en estos enfermos, sin dejar de tener presente, que:
Con el tiempo , todas las ictericias se aclaran.
Agradecimientos
A nuestros compañeros del Servicio de Gastroenterología, Endoscopía Digestiva y Unidad de Hepatología, por la enseñanza que fomentan en nosotros, durante su quehacer día a día.
Ala Señora Mirta Miranda Bonilla, por su colaboración siempre cordial y por su constante estímulo para que nos promovamos académicamente.
We present an up-to-date review article on the metabolism of bilirrubin, the causes of hiperbilirrubinemia, a classification of these and the management of patients with hepatobiliary disease and jaundice, according to the guidelines we apply optimizing our resources in our hospital.
Referencias
1. Lidofsky S y Scharschmidt BF. Ictericia. En: Sleissenger y Fordtran (ed) Enfermedades Gastrointestinales y Hepáticas. (6° ed). Buenos Aires: (Editorial Médica Panamericana) 2000. [ Links ]
2. Martin P y Friedman LS. Assesment of liver function and diagnostic studies. En: Friedeman LS, Keeffe EB (ed) Handbook of Liver Disease. (1º ed) Livingstone. Filadelfia: Churchill Livingstone. 1999. [ Links ]
3. Sherlock S. Diseases of the Liver and Biliary System (7 ed). Oxford: Blackwell Scientific Publications. 1985. [ Links ]
4. Vennes, JA y Bond JH. Approach to the jaundiced patient. Gastroenterology 1983; 84:1615-1618. [ Links ]
5. Guyton AC y Hall, JE. Textbook of Medical Physiology. (9 ed). Filadelfia: McGraw-Hill. 1998. [ Links ]
6. Berk PD y Noyer C. Billirubin metabolism and the hereditary hyper-bilirubinemias. Semin Liver Dis. 1994; 14: 321. [ Links ]
7. Kullac-Ublick GA, Hagenbuch B, Stieger B. Functional characterization of the basolateral rat liver organic anion transporting polypeptide. Hepatology 20: 411, 1994. [ Links ]
8. Sanyal AJ y Friedman LS. Evaluation of the Asymptomatic Patient with Abnormal LFT´s. En: AGA Spring Postgraduate Course Syllabus. San Francisco. 2002. [ Links ]
9. Te Boekhorst T, Urlus M, Doesburg W y otros. Etiologic factors of jaundice in severely ill patients. J Hepatol. 1988; 7: 111. [ Links ]
10. Pratt DS y Kaplan MM. Evaluation of abnormal liver-enzyme results in asymptomatic patients. N Engl J Med . 2000; 342 : 1266-71. [ Links ]
11. Brenner BM y Hebert SC. Trastornos de la función renal. En: Harrison Principios de Medicina Interna. (13 ed). Madrid, Mc-Graw Hill Interamericana 1994. [ Links ]
12. Alter MJ y Mast EE. The epidemiology of viral hepatitis in the United States. Gastroenterol Clin North Am. 1994; 23: 437. [ Links ]
13. Saini S. Imaging of the hepatobiliary tract. N Engl J Med. 1997; 336: 1889-94, 1997. [ Links ]
14. Bravo AA, Sheth SG y Chopra S. Liver biopsy. N Engl J Med. 2001; 344: 495-500. [ Links ]
15. Baron RL, Stanley RJ, Lee JKT y otros. A prospective comparison of the evaluation of biliary obstruction using computed tomography and ultrasonography. Radiology, 1982; 145: 91. [ Links ]
16. Goldberg HI, Korobkin MT. CT, ECHO before PTC, ERCP. Gastroenterology 1976; 72: 190-91. [ Links ]
17. Matzen P, Haubek A, Holst-Christensen J y otros. Accuracy of cholangiography by endoscopic or transhepatic route: A prospective study. Gastroenterology. 1981; 81-237-. [ Links ]
18. Kamath PS. Clinical approach to the patient with abnormal liver test results. Mayo Clin Proc 1996; 71: 1089-1095. [ Links ]Servicio de gastroenterología,
Hospital México, CCSS.
Abreviaturas: Br, bilirrubina; Hb, hemoglobina; Fe++, hierro; UDP, urudina difosfato; VGT,UDP gluconil transferasa; CMV, citomegalovirus; FA, fosfatasa alcalina; GGT, gama glutamil transpeptidona; ALT, alaminoramino transferasa,TP, tiempo de protombina.
Correspondencia:
luisdaniel@racsa.co.cr
hzamora13@hotmail.com
Unidad Académica de Hepatología y Servicio de Gastroenterología y Endoscopía Digestiva, Hospital México, CCSS-UCR * Universidad de Costa Rica, Escuela de Medicina

