











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introduction
Dental agenesis is a condition related to the congenital absence of teeth, and represents the most common anomaly of craniofacial development in humans (1). This condition can lead to dysfunctions in mastication, speech alterations, esthetic problems, and malocclusion (1). When dental agenesis occurs as an isolated trait or as a unique clinical finding, non-syndromic dental agenesis (NSDA) is diagnosed and appears either sporadically or in a familial fashion within a pedigree (1). Agenesis is classified according to the number of missing teeth as hypodontia (fewer than six missing teeth excluding third molars), oligodontia (six or more missing teeth excluding third molars), and anodontia (absence of all teeth) (2). The prevalence of NSDA has been estimated at 1.6%-36.5% depending on the studied population (3). Hypodontia, the most frequent dental abnormality, occurs in 3%-10% of cases, whereas oligodontia has a prevalence of 0.14%-0.5% (3). The teeth that are missing most frequently depend on the studied population: Agenesis of lateral incisors and second premolars is the most prevalent pattern (4), but for Asians, agenesis of lower incisors is predominant (3). Agenesis might be present in both the primary and permanent dentitions, and females are at a higher risk of being affected than males (3).
To date, more than 198 mutations in 15 genes have been related to NSDA etiology; however, 92% of these mutations are concentrated on seven genes: EDA, LRP6, WNT10A, WNT10B, PAX9, MSX1, and AXIN2 (5). Mutations in the last three genes-which encode the transcription factors muscle segment homeodomain-homeobox 1 (MSX1; 4p16.3-p16.1), paired box 9 (PAX9; 14q13.3), and axis inhibition protein 2 (AXIN2; 17q21-25)-have received the most research attention. The MSX1 and PAX9 genes encode homeodomain transcription factors expressed in the mesenchyme during the initial stages of dental development in response to epithelial signals (4).
The first gene identified as causative of NSDA was MSX1, a member of the homeobox transcription factor family with two exons (5). The MSX1 gene co-determines the position and shape of teeth, which involves the so-called homeobox code model, linking patterning of tooth types to the spatially restricted expression of homeobox genes in the dental mesenchyme (6). The PAX9 gene comprises five exons; it encodes an octapeptide, a pair of box domains, and a 128 amino acid paired homeodomain crucial in dental mesenchyme at all stages of odontogenesis (7). PAX9 is responsible for the expression of bone morphogenetic protein 4 (BMP4) that further regulates MSX1, thus evidencing its vital role in early tooth development stages (7). In addition, PAX9 has been suggested to establish the moment and place at which odontogenesis begins (7). Mutations in both PAX9 and MSX1 have been linked to NSDA, particularly to oligodontia (8-11). AXIN2, with 15 exons, plays an essential role as an intracellular inhibitor of the wingless-integrated (Wnt)/β-catenin signaling pathway; it is expressed in the enamel knot and mesenchymal odontoblasts during tooth formation (5). A mutation in AXIN2 from an oligodontia family evidenced the genetic link between NSDA and the Wnt pathway (12). Mutational analysis in two Mexican families living in Texas, USA, revealed the presence of 11 genetic variants: seven in AXIN2, three in PAX9, and two in MSX1, with one novel MSX1 mutation in pediatric cases with non-syndromic oligodontia (13). This case report suggests that a combined reduction in the MSX1 and PAX9 gene dosages increases the risk of oligodontia and supports the notion that tooth development requires an interaction between these genes (13).
In previous work by our research group, in Mestizo patients from Yucatan, Mexico (southeastern Mexico), the NSDA prevalence was 5.82% (n=670), with an approximate male-to-female ratio of 1:2 (14). The genetic background in that region comes from an admixture of European (39%), Amerindian (i.e., the Mayan ancestry (59%)), African (0.8%), and Asian (1.2%) genomes (15), which also might have resulted in the genetic variation in PAX9, MSX1, and AXIN2. The present study aimed to determine the genetic variants of PAX9, MSX1, and AXIN2 in Mayan probands with NSDA from Yucatan, Mexico.
Methods
Probands
We recruited seven patients of Mayan ethnicity with familial NSDA attending the School of Dentistry’s orthodontic clinic at the Autonomous University of Yucatan and private orthodontic clinics in Merida, Yucatan, Mexico (southeastern Mexico). The genetic ancestry of the Yucatan population has been reported previously; the main component is the Mayan ancestry (16, 17). Probands were selected if they had a familial history of NSDA based on their genealogy (with two or more affected first- or second-degree relatives) and with the diagnosis made by a trained orthodontist through clinical examination and inspection of panoramic radiographs. In addition, a certified geneticist evaluated the probands to exclude those with a clear syndromic presentation. Moreover, patients had to have involvement of teeth other than the third molars. Written informed consent was obtained from participants
>18 years old; younger participants were informed and their parents provided written consent. Cases with incomplete data, defective panoramic radio- graphs, clinically identifiable syndromes, and cleft lip/palate were excluded. At the time the research was conducted, none of the probands had been diagnosed with colorectal cancer, other neoplasia, or additional clinical phenotypes. The study proto- col was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the Ethics Committee of Autonomous University of Yucatan.
Genotyping of msx1, pax9 and axin2 variants
Genomic DNA was extracted from peripheral blood by using the Quick-gDNA™ MiniPrep Extraction Kit, according to the protocol established by the supplier.
DNA was amplified with polymerase chain reaction (PCR) in a final volume of 15µL with the DreamTaq polymerase and previously described primers for specific exons and functional domains of MSX1 (exon 2, encoding the homeodomain) and PAX9 (exons 2-4, encoding the paired domain) (18), and AXIN2 (exons 2b, 6, and 7, encoding the regulator of G protein signaling (RGS) domain) (13). Exon 2 of MSX1 was amplified with the following PCR conditions: 95ºC for 2min; 35 cycles of 95ºC for 30s, 52ºC for 45s, and 72ºC for 1min; and a final extension at 72ºC for 5min. Exons 2-4 of PAX9 were amplified as follows: 95ºC for 2min; 35 cycles at 95ºC for 30s, 57.3ºC (exon 2) or 49.7ºC (exon 3) or 51.8ºC (exon 4) for 45s, and 72ºC for 1min; and a final extension at 72ºC for 5min. For AXIN2, the PCR conditions were: 95ºC for 2min; 35 cycles at 97ºC for 40s, 56.6-57ºC (exon 2b) or 57.5-58ºC (exon 6) or 55.6-58ºC (exon 7) for 45s, and 72ºC for 1min; and a final extension at 72ºC for 5min.
The PCR products were visualized in 2% agarose gels stained with ethidium bromide. Then, the amplified fragments were purified with the commercial Zymoclean Gel DNA Recovery Kit according to the manufacturer’s protocol. The amplicons were quantified with a Promega Quantus Fluorometer and the QuantiFluor One dsDNA system. Finally, the quality of the products was verified based on amplification of the housekeeping gene methyle- netetrahydrofolate reductase (MTHFR), assessed in 2% agarose gels.
Genetic variants were screened with bidirectional Sanger sequencing using the BigDye Terminator v3.1 cycle sequencing kit, the Themocycle Arktik Thermal Cycler from Thermo Scientific, and the ABI Prism 310 capillary sequencer. The resultant fragments were purified with the BigDye X Terminator Kit. Chromatograms were visualized and aligned to the reference sequences of targeted regions from the human genome assembly GRCh38.p13 with the MEGA 11 software.
Bioinformatic analysis
Pairwise global alignment was performed with the Geneious Alignment program (Geneious v8.1.9) using the forward and reverse sequences of each sequenced exon. The 5' and 3' ends of each sequence that did not match in alignment were eliminated; only the sequences that matched were considered for further analysis. Basecalling and decomposition were developed by using the forward and reverse Sanger Chromatogram trace files (ab1 format) to improve variant calling and utilizing the reference genome GRCh38.p13 with the command-line application Tracy v0.5.5. The resulting files were normalized, and forward and reverse files (binary variant call format (BCF)) were merged and converted to the variant call format (VCF) with bcftools v1.9. The variant annotations and predicted effects were determined by using the online programs Variant Annotation Integra- tor (https://genome.ucsc.edu/cgi-bin/hgVai) and Variant Effect Predictor (https://www.ensembl.org/ Tools/VEP), respectively, with the Sorting Intolerant from Tolerant (SIFT) and the Polymorphism Phenotyping v2 (PolyPhen-2) algorithms. The pathogenic, likely pathogenic, benign, likely benign, or uncertain significance category was assigned according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) criteria (19). Finally, four genomes from unaffected persons with Mayan ancestry (HGDP00854, 00856, 00858, and
00859; https://www.internationalgenome.org/ data-portal/population/MayaHGDP) were included for comparative purposes only.
Results
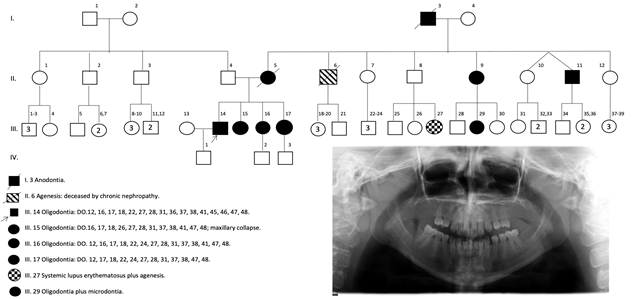
Six of the seven probands were adults (19-33 years old), and one was an adolescent (12 years old); according to sex, six of the probands were female (Table 1). Two probands (1 and 1.1), whose representative pedigree is depicted in Figure 1, were siblings. The pedigrees from probands 2-6 are depicted in Figures S1-S5. NSDA was observed in deciduous teeth and exhibited a dominant inheritance pattern (except for families 3 and 4).
According to the agenesis classification, only probands 1 and 1.1 were diagnosed with oligodontia (>6 missing teeth, excluding the third molars), whereas probands 2-6 had hypodontia. Proband 1 exhibited the most evident phenotype with 16 missing teeth (seven from the upper jaw and nine from the lower jaw). In addition, the pedigree from this proband was characterized by several cases of oligodontia and even one patient with anodontia (a deceased grandmother without additional clinical data); other diseases such as systemic lupus erythematosus or chronic nephropathy were also observed. Only one sister of proband 1 (proband 1.1) agreed to be included in the study. She also had oligodontia with a very similar pattern of missing teeth as proband 1, with the exception of dental organs 24, 36, 46, and 46 (Table 1). Despite our efforts, no other sibling or parent from other probands agreed to be included in the study or they were inaccessible, and some relatives were deceased.
Table 1 depicts the variants in specific exons from the PAX9, MSX1, and AXIN2 genes; zygosity; the reported minor allele frequency (MAF); the clinical significance according to the ACMG/AMP; and the dental organ(s) affected. Five probands showed variants in the AXIN2 gene, two probands showed variants in PAX9, and two probands showed a 3' untranslated region (UTR) variant in the MSX1 gene. Both jaws of probands 1 and 1.1 had missing teeth, but they only carried a genetic variant in exon 3 of PAX9. Probands 2 and 6 had missing teeth in the upper jaw, with variants only in the AXIN2 gene. The lower jaw of probands 3 and 4 was affected, and they had variants in AXIN2 and MSX1. Finally, proband 5 also had missing teeth in the lower jaw, but the variants were localized in PAX9 and AXIN2.
As noted, AXIN2 showed the highest number of variants, particularly in exon 6. These variants have previously been described and classified according to their clinical significance into variants with uncertain significance (VUS) (rs1060502133, rs1060502139, rs147716924, rs1330822418, rs769741903), likely benign (rs9915936), and benign (rs1133683). Of note is rs1234437759 (G>A, exon 7). It is not reported on the ClinVar website, and according to the ACMG/AMP criteria (19), the change is predicted to be “benign” based on the SIFT (result=tolerant) and PolyPhen-2 (result=benign) algorithms. Furthermore, the generated fragments encompassed segments from introns 7-8, thus identifying the benign intronic variants rs28760438 and rs9906513.
Regarding PAX9, the genetic variants were in exon 3 (i.e., the benign variants rs12881240 and rs4904210), whereas there were no variants in exons 2 and 4. Interestingly, probands 1 and 1.1 showed variants in exon 3 of PAX9, although the former had rs12881240 and the latter had rs4904210. On the other hand, rs8670 was the only mutation found in the 3' UTR region of the MSX1 gene; the reported clinical significance is benign.
Because no additional relatives of probands were able to participate in the study, we compared our results against reference genomes of four unaffected individuals of Mayan ancestry (Table 1). There was no variant in the MSX1 gene, whereas PAX9 had rs4904210 (exon 3) and AXIN2 had rs9915936 (exon 6), rs1133683 (exon6), rs28760438 (exon 7), and rs9906513 (exon 7).
Table 1 Missing teeth and mutations in PAX9, MASX1, and AXIN2 according to the patient characteristics.
| Genes, exons, and variants* | - | - | - | - | - | - | - | - | - | - | - | - |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proband | Sex | Age | Type of agenesis | MSX1 | PAX9 | - | - | AXIN2 | - | Missing teeth | - | |
| - | - | - | - | Exon 2 | Exon 2 | Exon 3 | Exon 4 | Exon 2b | Exon 6 | Exon 7 | Upper jaw | Lower jaw |
| 1 | M | 33 | Oligodontia | --- | --- | rs12881240 | --- | --- | --- | --- | DO: 12, 16, | DO: 31, 36, |
| - | - | - | - | - | - | (G/A; 0.13; B) | - | - | - | - | 17, 18, 22, | 37, 38, 41, |
| - | - | - | - | - | - | - | - | - | - | - | 27, 28 | 45, 46, 47, |
| - | - | - | - | - | - | - | - | - | - | - | - | 48 |
| 1.1 | F | 30 | Oligodontiaw | --- | --- | rs4904210 (G/C; 0.37; B) | --- | --- | --- | --- | DO: 12, 16, 17, 18, 22, | DO: 31, 37, 38, 41, 47, |
| - | - | - | - | - | - | - | - | - | - | - | 24, 27, 28 | 48 |
| 2 | F | 27 | Hypodontia | --- | --- | --- | --- | --- | rs1133683 | rs28760438 | DO: 12 | Unaffected |
| - | - | - | - | - | - | - | - | - | (G/A; 0.60; B) | (A/G; 0.57; B)† | - | - |
| - | - | - | - | - | - | - | - | - | rs9915936 | rs9906513 | - | - |
| - | - | - | - | - | - | - | - | - | (T/C; 0.90; LB) | (A/T; 0.04; B)† | - | - |
| 3 | F | 19 | Hypodontia | rs8670 (C/T; 0.25; B) | --- | --- | --- | --- | rs1133683 | rs28760438 | Unaffected | DO: 45 |
| - | - | - | - | - | - | - | - | - | rs9915936 (C/C; | - | - | - |
| - | - | - | - | - | - | -- | - | - | 0.90; LB) | - | - | - |
| - | - | - | - | - | - | - | - | - | rs1060502133 | - | - | - |
| - | - | - | - | - | - | - | - | - | (A/G; 0.0; US) | - | - | - |
| - | - | - | - | - | - | - | - | - | rs147716924 | - | - | - |
| - | - | - | - | - | - | - | - | - | (G/A; < 0.001; US) | - | - | - |
| - | - | - | - | - | - | - | - | - | rs1330822418 | - | - | - |
| - | - | - | - | - | - | - | - | - | (T/C; 0.0; US) | - | - | - |
| - | - | - | - | - | - | - | - | - | rs1060502139 | - | - | - |
| - | - | - | - | - | - | - | - | - | (G/A; < 0.001; US) | - | - | - |
| -- | - | - | - | - | - | - | - | - | rs769741903 | - | -- | - |
| - | - | - | -- | - | -- | (T/C; 0.0; US) | - | - | - | |||
| 4 | F | 29 | Hypodontia | rs8670 (C/T; 0.25; B) | --- | --- | --- | rs2240307 (A/G; 0.013; B) | rs1133683 (G/A; 0.60; B) rs9915936 (T/C; 0.90; LB) | rs9906513 (A/T; 0.04; B)† rs28760438 (A/G; 0.57; B)† | Unaffected | DO: 35, 45 |
| 5 | F | 12 | Hypodontia | --- | --- | rs12881240 (G/A; 0.13; B) | --- | --- | --- | rs1234437759 (G/A; 0.0; NR)‡ | Unaffected | DO: 41 |
| - | - | - | - | - | - | rs4904210 (G/C; 0.37; B) | - | - | - | rs28760438 (G/G; 0.57; B)† | -- | - |
| 6 | F | 20 | Hypodontia | --- | --- | --- | --- | --- | rs9915936 (C/C; 0.90; LB) | --- | DO: 15, 25 | Unaffected |
| HGDP 00854§ | F | Unaffected | --- | --- | rs4904210 (G/C; 0.37; B) | --- | --- | rs9915936 (C/C; 0.90; LB) | rs28760438 (A/G; 0.57; B)† | --- | --- | |
| - | - | - | - | - | - | - | - | - | rs1133683 (G/A; 0.60; B) | rs9906513 (A/T; 0.04; B)† | - | - |
| HGDP 00856§ | M | --- | Unaffected | --- | --- | --- | --- | --- | rs9915936 (C/C; 0.90; LB) | rs28760438 (A/G; 0.57; B)† | --- | --- |
| - | - | - | - | - | - | - | - | - | rs1133683 (A/A; 0.60; B) | rs9906513 (A/T; 0.04; B)† | - | - |
| HGDP 00858§ | F | --- | Unaffected | --- | --- | rs4904210 (G/C; 0.37; B) | --- | --- | rs9915936 (C/C; 0.90; LB) | rs28760438 (G/G; 0.57; B)† | --- | --- |
| - | - | - | - | - | - | - | - | - | rs1133683 (A/A; 0.60; B) | - | - | - |
| HGDP 00859§ | F | --- | Unaffected | --- | --- | rs4904210 (G/C; 0.37; B) | --- | --- | rs9915936 (C/C; 0.90; LB) | rs28760438 (G/G; 0.57; B)† | --- | --- |
| - | - | -- | --- | - | - | - | - | rs1133683 (A/A; 0.60; B) | - | - | - |
*Each variant is presented with its respective rsID number. Zygosity, the minor allele frequency (from the ALFA project: https://www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/), and the ClinVar website clinical significance according to the American College of Medical Genetics and Genomics (ACMG) (NR, not reported; US, uncertain significance; LB. likely benign; and B, benign) are in brackets.
†Variants were found in intronic regions.
‡rs124437759 is not reported on the ClinVar website. However, the predicted effect is “benign” from the ACMG criteria based on the Sorting Intolerant from Tolerant (SIFT) (result = tolerant) and Polymorphism Phenotyping v2 (PolyPhen-2) (result = benign) algorithms.
§Reference genomes from people with Mayan ancestry extracted from the International Genome Sample Resource (https://www.internationalgenome.org/data-portal/population/MayaHGDP). Abbreviations: DO, dental organ; M, male; F, female.
Discussion
This study is the first, to our knowledge, to describe the genetic variants of PAX9, MSX1, and AXIN2 in patients of Mayan ancestry with familial NSDA from southeastern Mexico. All probands showed genetic variants in AXIN2, particularly in exons 6 and introns 7-8. Additionally, three probands exhibited mutations in exon 3 of the PAX9 gene, while only two probands carried a variant in the 3' UTR region of the MSX1 gene. Moreover, some of these variants were present in the genomes of healthy subjects of Mayan ancestry.
The AXIN2 gene exhibited the highest number of variants, particularly in exon 6 and introns 7-8 in almost all probands. Similarly, Haddaji Mastouri et al. (4) found that the majority of the genetic variants in an NSDA case series were present in the AXIN2 gene. AXIN2 encodes an intracellular inhibitor of the Wnt/β-catenin signaling pathway and is mainly expressed in the enamel knot and mesenchymal odontoblasts during odontogenesis (5). Mutations in AXIN2 have been reported in familial oligodontia and anodontia. Conversely, in our study, proband 1 and his sister (proband 1.1) showed the highest number of missing teeth, 16 and 14, respectively, with involvement of the upper and lower third molars. However, they did not carry AXIN2 variants. The other probands carried known single nucleotide polymorphisms (SNPs) in exon 6: rs1133683, classified as benign by the ClinVar website, is associated with non-syndromic cleft lip (20) and colorectal cancer (21), but not with NSDA; rs9915936, classified as benign in ClinVar, is associated with cancer risk (22), but not with NSDA; and rs1060502133, rs147716924, rs1330822418, rs1060502139,
and rs769741903 have uncertain significance for oligodontia and cancer risk.
On the other hand, there were two benign SNPs in AXIN2 introns 7-8, namely rs28760438 and rs9906513. Furthermore, we found rs1234437759 as a heterozygous G>A change; it is not reported on the ClinVar website. According to the ACMG/ AMP criteria (19), the change is predicted to be benign based on the SIFT (result=tolerant) and PolyPhen-2 (result=benign) algorithms. Recently, novel variants of AXIN2 have been described in other exons, including exon 2 (rs752881223) (23), exon 3 (rs77908340) (24) and exon 11 (rs747010025) (24) in Chinese families. In Mexican families with a predominantly oligodontia phenotype (7-14 missing teeth), rs2240398 (exon 2b), rs35399989 (introns 4-5), c.1201+69A>G (intron 5), rs9915936 (exon 6; also observed in probands 2, 3, 4, and 6 in our study), rs1133683 (exon 6; also observed in probands 2, 3, 4, and 6 of our study), c.1907+73T>C (intron 7-8), and c.1904-18C>G (introns 7-8) have been reported (13).
To date, the phenotype-genotype correlation in carriers of AXIN2 variants is unclear. For example, Swedish carriers of the pathogenic SNPs rs138287857 (exon 8) and rs145007501 (exon 10) were found with 7 and 10 missing teeth, respectively (25). Moreover, rs779083840 (exon 3) and rs747010025 (exon 11) produced a phenotype of 12 missing teeth in Chinese carriers. rs752881223 (exon 2) was recently found in a Chinese proband with 7 missing teeth (23). By contrast, our probands 1 and 1.1 exhibited 16 and 14 missing teeth, respectively, without known or novel variants in exons 2b, 6, or 7 of AXIN2. Probands 2-6 showed different patterns of known variants in the three exons, with one (probands 2, 3, and 5) or two (probands 4 and 6) missing teeth in the upper jaw (probands 2 and 6) or lower jaw (probands 3, 4, and 5). These findings are consistent with the study by Haddaji Mastouri et al. (4). who suggested that hypodontia and oligodontia are both associated with mutations in the AXIN2 gene. Additionally, for our probands with familial NSDA, we cannot discard an interaction between defective AXIN2 and other affected proteins, such as PAX9.
PAX9 is a well-known transcription factor that regulates the expression of other key mesenchymal odontogenic proteins during tooth development at the bud stage (5). Mutations in PAX9 exon 3 are of particular interest because this region encodes the paired DNA-binding domain that confers its transcription factor activity. Most genetic variants found in our probands with familial NSDA are localized in exon 3 of PAX9. These genetic variants might affect the DNA-binding domain and increase the regulatory function of the protein, given that exon 3 represents the largest coding sequence of PAX9. Mutations in such an essential region may lead to a loss of transcriptional activity (7). For example, g.10672A>T has recently been found as a pathogenic variant in exon 3. Additional mutations include c.146delC, c.218dupG, c.256_262dup, c.76C>T, and C.140G>C, among others (8). In the present study, we found rs12881240 (26,27) (exon 3, probands 1 and 5) and rs4904210 (27,28) (exon 3, probands 1.1 and 5) that have previously been reported to be associated with NSDA. In Mexican families, rs12881240 and rs4904210, both in exon 3, have been reported; they impact protein and messenger RNA (mRNA) structures (13). In particular, rs4904210 has been suggested as a neutral polymorphism that is highly conserved among humans and primates. Therefore, such a genetic variant could represent an evolutionary window to functional adaptations in the primate dentition (29). We observed that most of our probands with genetic variants in PAX9 were missing molars in the lower jaw, including a proband and his sibling (probands 1 and 1.1) with the most missing teeth. Regarding the pattern of missing teeth, Ren et al. (30) found that PAX9 genetic variants predominantly lead to the absence of second molars in the following order: missing upper second and first molars, upper second premolars, and lower second molars.
Only probands 3 and 4 carried the heterozygous rs8670 in the 3' UTR of MSX1 and presented one (dental organ 45) and two (dental organs 33 and 45) missing premolars, respectively. The MSX1 gene encodes a homeobox transcription factor involved in the early stages of tooth development. It is a relevant gene for establishing tooth shape and position (31); its mutations are associated with missing premolars and particular syndromic conditions such as cleft lip/palate and Witkop syndrome (32, 33). Haddaji Mastouri et al. (4) showed that the majority of patients with MSX1 variants presented exclusive agenesis of second premolars, or in combination with incisors, and agenesis of the canines. A Chinese family with a non-stop mutation in the MSX1 gene also showed the typical pattern in the proband lacking all four second premolars, two maxillary first premolars, and four third molars (6). Some mutations related to dental agenesis are rs36059701 (Czech cases) (9), rs121913130 (American cases) (34), rs515726227 (Chinese cases) (6) and rs8670 (Saudi Arabian cases) (10). In Mexican families, MSX1 c.476T>G was demonstrated to change the protein and mRNA structures and has been linked to oligodontia (13).
We also compared our results against four reference genomes of unaffected people with Mayan ancestry. None of the genomes carried variants in the MSX1 gene. Moreover, the only SNP found in PAX9 was the heterozygous rs4904210. Interestingly, probands 1 (16 missing teeth) and 1.1 (14 missing teeth) carried rs12881240 and rs4904210, both associated with NSDA in epidemiological studies (26,27) and observed in mutational screening studies (35). Furthermore, the reference Mayan genomes also had homozygous rs9915936 (MAF 0.90) and homozygous or heterozygous rs1133683 (MAF 0.6), rs28760438 (MAF 0.57), and rs9906513 in AXIN2. These results suggest that some variants in the three evaluated genes might be present in the overall population, but others might be epidemiologically associated with NSDA. We cannot exclude the presence of variants in other exons of MSX1, PAX9, and AXIN2, as well as in WNT10A or genes that encode members of the EDA/EDAR/NF-kB signaling pathway (36). Moreover, it is possible that synergic effects with other genes involved in tooth development lead to familial NSDA (37).
Our study had some limitations. For example, we could not sequence non-affected siblings of the probands due to their refusal to participate in the study, our inability to contact them, or the fact that some of them were deceased. Moreover, we only evaluated PAX9, MSX1, and AXIN2, thus limiting the implication of additional genes such as FGRF1, EDA, EDAR, LRP6, and WNT10A, among others (2,36). Hence, future whole-genome/exome sequencing studies in larger cohorts and controls will allow the identification of novel loci and deleterious mutations that might provide insight into the complete genetic mechanisms involved in dental agenesis.
CONCLUSION
AXIN2 accounted for the highest number of genetic variants, with rs1060502133, rs1060502139, rs147716924, rs1330822418, rs769741903, rs9915936, rs1133683, and rs1234437759. We also found two variants in PAX9 (rs12881240 and rs4904210) and one in MSX1 (rs8670). Because some of these SNP were present in genomes from unaffected people of Mayan ancestry, additional functional and epidemiological studies are required to evidence the mutational patterns associated with a specific phenotype. Moreover, future whole-genome/exome sequencing analyses in these cases and healthy controls are required to accurately characterize the underlying genetic mechanisms of dental agenesis.
Abbreviations
ACMG, American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; BCF, binary variant call format; MAF, minor allele frequency; NSDA, non-syndromic dental agenesis; PCR, polymerase chain reaction; VCF, variant call format; PolyPhen-2, Polymorphism Phenotyping v2; SIFT, Sorting Intolerant from Tolerant; VUS, variant with uncertain-significance.
Ethical statement
Written informed consent was obtained from participants >18 years old; younger members were informed about the study, and their parents provided written consent. The study protocol was conducted according to the principles expressed in the Declaration of Helsinki and approved by the Ethics Committee of the Autonomous University of Yucatan.
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
Data availability statement
The datasets presented in this article are not readily available because ethical approval for the underlying studies prohibit sharing of data. Requests to access the datasets should be directed to Dr. Herrera-Atoche (jose.herrera@correo.uady.mx) or Dr. Gonzalez-Herrera (lizbeth@correo.uady.mx).
Funding
This work was supported by institutional funds from the School of Dentistry of Autonomous University of Yucatan (FODO-2015-0001) and from DIMYGEN Lab. S.C.P.
Author contribution statement
Evaluated patients and obtained samples: N.A.G.P. and J.E.C.M.
Contributed to the conception and design of the study: L.G.H. and J.R.H.A.
Performed the Sanger-sequencing experiments: P.L.G.B. and J.E.S.E.
Provided resources and supervised the clinical evaluation of patients: F.J.A.A. and I.D.Z.H.
Wrote the first draft of the manuscript: J.A.R.M. Performed the bioinformatic analysis: J.A.R.M. and R.P.A.

