











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introducción
Los principales países productores de soja son: Brasil (39 %), Estados Unidos (29 %), Argentina (13 %), China (5 %) y Paraguay (3 %). Entre los exportadores de granos, harinas, aceites y biocombustibles se destacan Brasil (61 %), Estados Unidos (29 %), Paraguay (3,9 %) y Argentina (2,8 %) (United States Department of Agriculture, 2024). Enfermedades como roya asiática, mancha marrón, diferentes tizones, cancro y muerte súbita afectan la calidad de semillas, granos y derivados, y generan pérdidas de rendimiento cercanas a 11 % (Allen et al., 2017). El uso de semilla de mala calidad, el monocultivo de soja y la presencia de residuos de cosecha en superficie son algunos de los factores que favorecen la supervivencia del inóculo y la ocurrencia de enfermedades en el siguiente ciclo de cultivo (Peruzzo et al., 2019).
El cancro del tallo de la soja (CTS) se caracteriza por el desarrollo de una necrosis deprimida en el tallo, causada por dos variedades del hongo Diaporthe phaseolorum (fase sexual o teleomorfo de Phomopsis phaseoli), determinadas como D. phaseolorum var. meridionalis (Dpm) y D. phaseolorum var. caulivora (Dpc). En Argentina, la aparición del CTS causado por Dpm (CTS-Dpm) ocurrió en el año 1992 en lotes de las provincias de Buenos Aires y Santa Fe, con valores de incidencia (% I) entre 5 y 8 %, para 1997 este parámetro alcanzó valores entre 70 y 100 %. El primer reporte de CTS causado por Dpc (CTS-Dpc) ocurrió en 2001 en regiones de Santa Fe, Córdoba y Buenos Aires (Pioli et al., 2023).
La resistencia al CTS se sustentó originalmente en cuatro genes de resistencia identificados y reportados como Rdc (de Rdc1 a Rdc4). Sin embargo, la revisión exhaustiva realizada sobre antecedentes de la CTS, el origen geográfico de los aislamientos (Diaporthe phaseolorum “sureño o meridionalis”) utilizados como inóculo y el comportamiento biológico y patogénico en Estados Unidos (1987-1996), validado posteriormente con interacciones comparables en Argentina (1997-2003), permitió identificarlos morfológica y molecularmente como aislamientos de Dpm que causaron la enfermedad CTS-Dpm. Por tal motivo, Pioli et al. (2003) redenominaron aquellos genes Rdc1-4 como genes Rdm1-4 de resistencia a CTS-Dpm, los cuales además no eran efectivos para CTS-Dpc.
En consecuencia, la variedad (var.) Tracy-M resultó ser portadora de Rdm1 y Rdm2, la var. Crockett fue portadora del gen Rdm3, y las var. Dowling y Hutcheson presentaron el gen Rdm4. Posteriormente, Chiesa et al. (2013) identificaron el gen Rdm5 en este último genotipo. Este avance permitió comprender la incorporación extendida de la var. Hutcheson como fuente de resistencia (R) a CTS-Dpm en programas de mejoramiento de soja en Estados Unidos y Argentina (Chiesa et al., 2017). La prevalencia de CTS-Dpc fue consecuencia de la carencia de genotipos portadores de genes de R a CTS-Dpc y la disponibilidad predominante de germoplasma de soja con genes Rdm, efectivos para CTS-Dpm pero no para CTS-Dpc (Pioli et al., 2023).
A nivel regional, en los años 2001 y 2006 se registraron el primer reporte de CTS-Dpc en Argentina y Rio Grande Do Sul, Brasil, respectivamente. Nuevos registros de CTS-Dpc fueron reportados partir de 2002, en la zona productiva de Argentina, así como la infección de CTS causada por Dpm y Dpc en Uruguay (Pioli et al., 2019). Las evaluaciones moleculares de genotipos comerciales de soja demostraron, en las últimas décadas, que éstos son mayoritariamente portadores de genes Rdm.
La incorporación y análisis de la herencia de la resistencia a CTS-Dpc se realizó mediante un análisis mendeliano clásico, que a través de la caracterización fenotípica de las familias F2:3 (prueba de progenie) identificó el primer gen en la población segregante (PS): PS-258 (GP13 x GP4, donde GP refiere a genotipo progenitor). Este gen fue aportado por el GP13, que mostró R, y fue denominado Rdc1 (Peruzzo et al., 2019). En estudios ampliados a otras PS −PS-157 (GP13 x GP12), que comparte el progenitor GP13, y PS-172 (GP9 x GP16), cuyos progenitores son portadores de genes Rdm de resistencia a CTS-Dpm pero carecen de genes de resistencia conocidos a CTS-Dpc (López Achaval et al., 2014; Peruzzo, 2018)− se reportaron resultados que evidencian una herencia más compleja, ya que no ajustaron a una segregación mendeliana 3R:1S.
A partir de estas observaciones, surgió la necesidad de aplicar métodos de genética cuantitativa que permitan detectar tanto variaciones genotípicas como eventuales genes modificadores, con el fin de interpretar la herencia de este gen, reportado por el equipo de trabajo. La aplicación de enfoques cuantitativos clásicos, ampliamente conocidos y aplicados por los fitomejoradores para evaluar la resistencia a enfermedades de diferentes cultivos podría permitir, en este caso, la validación de los resultados obtenidos para resistencia a CTS-Dpc desde el enfoque de la genética mendeliana. También es posible estimar parámetros genéticos para definir estrategias de manejo de las poblaciones segregantes obtenidas.
El conocimiento de la heredabilidad de un carácter permite dilucidar qué proporción de la variación fenotípica de dicho carácter se debe a varianza en los factores genéticos y ambientales. El valor de la heredabilidad varía entre 0 a 1, donde un valor cercano a 0 indica que la variabilidad fenotípica en dicho carácter está mayormente condicionada por la variabilidad en los efectos del ambiente que determinan su expresión. Por el contrario, un valor cercano a 1 indica que la mayor parte de la variación del carácter se debe a variaciones en los efectos genéticos (Cabodevila et al., 2017).
La asociación de ciertos caracteres cuantitativos y su expresión fenotípica se conoce como correlación fenotípica. Ésta puede desglosarse en correlación genética y correlación ambiental. La primera resulta del ligamiento o la pleiotropía y la segunda responde a efectos ambientales que afectan simultáneamente la expresión de los caracteres asociados.
La correlación genética permite realizar selección indirecta y detectar respuestas correlacionadas no deseadas. La selección indirecta permite la selección de un determinado carácter “y”, que difícilmente puede ser medida por la complejidad que pueda resultar o por la imposibilidad de hacerlo en etapas tempranas, a través de otro carácter “x” de más fácil o más precoz medición o de mayor heredabilidad. La respuesta correlacionada, por su parte, puede provocar la selección de un carácter no deseado debido a la asociación de esos caracteres (Kearsey & Pooni, 2020). El objetivo de la investigación fue aplicar enfoques de la genética cualitativa y cuantitativa clásicas para la identificación del gen Rdc1 de resistencia a la cancrosis del tallo de soja causada por Dpc.
Materiales y métodos
Para lograr el objetivo planteado, se evaluó la herencia mendeliana mediante el método cualitativo y se determinaron el grado de dominancia, la heredabilidad de la resistencia a CTS-Dpc en generaciones tempranas y avanzadas, el número de genes de resistencia involucrados y las correlaciones fenotípica, genotípica y ambiental entre la incidencia y severidad desde el enfoque cuantitativo. Se seleccionó un germoplasma fúngico con interacciones compatibles en Glycine max. Se analizaron tres poblaciones segregantes obtenidas a partir de cinco genotipos progenitores, dos de las cuales compartieron el mismo progenitor portador del gen Rdc1, resistente a CTS-Dpc.
Selección del germoplasma fúngico
La inoculación de las poblaciones segregantes F3, completas y ordenadas en familias (F2:3) (pruebas de progenie), se realizó con el aislamiento Dpc16 (procedente de Esperanza, Santa Fe, Argentina). Este aislamiento fue validado morfológica y molecularmente y demostró ser capaz de diferenciar la reacción de resistencia (R) o susceptibilidad (S) de los genotipos progenitores en sucesivas evaluaciones experimentales (López Achaval et al., 2014; Peruzzo et al., 2019; Pioli et al., 2006; 2019). Las inoculaciones se realizaron de acuerdo con protocolos previamente aplicados y en trabajos anteriores, con una suspensión ajustada a aproximadamente 1,5×105 conidios/ml. La virulencia de Dpc16 fue preservada mediante inoculaciones periódicas de plantas susceptibles y posterior reaislamiento del patógeno desde las zonas infectadas y sintomáticas (tercer y cuarto postulados de Koch) (Hernández et al., 2015; Peruzzo et al., 2018).
Selección de genotipos progenitores de soja
Los progenitores se seleccionaron con base en el comportamiento frente a la CTS-Dpc durante los años 2012, 2013, 2017 y 2020. La CTS-Dpc se estimó a través de la incidencia (I %) (porcentaje de plantas infectadas / total de plantas evaluadas) y la severidad (S % = ∑ % área sintomática asignada a cada planta / número total de plantas evaluadas de cada genotipo) (Madden et al., 2007). El porcentaje de área sintomática en cada planta fue estimado con base en una escala de S de seis categorías o niveles de daño (Cuadro 1), la cual fue aplicada por Chiesa et al. (2017), López Achaval et al. (2014), Peruzzo et al. (2021) y Pioli et al. (2003).
Cuadro 1 Escala de severidad con seis niveles de daño o sintomatología de CTS-Dpc observada en plantas desde el momento de la inoculación hasta 49 días postinoculación, desarrolladas en el Campo Experimental J Villarino, Zavalla, Santa Fe, Argentina, durante los años 2012, 2013 y 2017.
| Escala de severidad1 | Significancia fenotípica |
| 02 | Sin síntomas. |
| >0 hasta 0,302 | Cancro en torno al punto de inoculación o elongado sin traspasar el nudo cotiledonar, hasta finalizar la evaluación (49 dpi). |
| O Moteado clorótico en hojas unifoliadas. | |
| >0,3 hasta 0,453 | Ambos síntomas: expansión del cancro al primer entrenudo del epicótilo y moteado, abigarrado o marchitez de una hoja trifoliada. |
| >0,45 hasta 0,603 | Ambos síntomas: cancro extendido en 2 entrenudos en epicótilo y marchitez acentuada con curvatura del ápice caulinar, pero el tallo preserva entrenudos superiores turgentes. |
| >0,60 hasta 0,753 | Ambos síntomas: cancro extendido en entrenudos del epicótilo y marchitez acentuada foliar y caulinar. |
| >0,75 hasta 13 | Necrosis irreversible o planta muerta. |
1Los valores expresados como proporciones permiten ser expresados en su correspondiente valor porcentual (%). / 1The values expressed as proportions can be expressed as corresponding percentage value (%).
2Valores escala: desde 0 y hasta 0,3 a los 49 dpi corresponden a plantas sanas o resistentes (PSR). / 2Scale values: From 0 to 0.3 at 49 dpi correspond to healthy or resistant plants (HRP).
3Valores escala >0,3 hasta alcanzar el valor 1 a los 49 dpi, corresponden a plantas enfermas o susceptibles (PES). / 3Scale values of >0.3 to 1 at 49 dpi correspond to diseased or susceptible plants (DSP).
El comportamiento fenotípico y sanitario diferencial de los cinco genotipos de soja de interés agronómico, seleccionados como progenitores de las tres poblaciones segregantes evaluadas, se muestra en el Cuadro 1. El GP13 es portador del gen Rdc1 efectivo para CTS-Dpc (Peruzzo et al., 2019) y el GP9 es portador de dos genes Rdm de resistencia a CTS-Dpm. El resto (GP4, GP12 y GP16) corresponden a cultivares comerciales de valor agronómico y con buen comportamiento frente a CTS-Dpm, pero sin genes Rdc previamente identificados (Pioli et al., 2023).
Selección de las poblaciones segregantes resultantes de cruzamientos de cinco progenitores de soja
Las poblaciones segregantes F2 seleccionadas para este estudio, PS-258 (GP13 x GP4), PS-157 (GP13 x GP12) y PS-172 (GP9 x GP16), derivaron de individuos F1 heterocigotas e híbridos, validados molecularmente mediante marcadores de polimorfismo de nucleótido único (SNP) (Peruzzo, 2018). En consecuencia, son poblaciones independientes, aunque dos de ellas comparten al padre resistente GP13. Los lotes de semillas, plantas F2 y poblaciones segregantes F3 seleccionadas fueron sembrados en invernadero. Las poblaciones F3 fueron conducidas como familias segregantes F2-3 e inoculadas cuando las plántulas estaban en la etapa vegetativa de la primera a la segunda hojas trifoliadas totalmente expandidas.
Las inoculaciones y el progreso de la enfermedad se evaluaron cada semana a partir de los siete días después de la inoculación y hasta los 49 días después de la inoculación (dpi) (Hernández et al., 2020; López Achaval et al., 2014; Peruzzo et al., 2019). La expresión fenotípica (síntomas) de CTS-Dpc fue evaluada con base en la incidencia (I %) de CTS-Dpc observada en cada familia F2-3 y la severidad (S %) de la enfermedad registrada en cada planta o individuo segregante dentro de cada familia.
A cada planta y genotipo se le asignó un valor de S % de acuerdo con la escala citada previamente (Cuadro 1). Aquellas plantas con valores de S % de 0 y 0,3 fueron consideradas plantas resistentes (PR) y las que presentaron valores superiores a 0,3 y alcanzaron un valor igual a 1 (planta muerta, PM) se consideraron plantas enfermas y susceptibles (PES) (Peruzzo et al., 2019). Estos valores de proporción pueden expresarse como sus correspondientes porcentajes (%), sin afectar el significado biológico.
Las semillas F4 derivadas de la PS-258 (GP13 x GP4) fueron sembradas; sin embargo, los individuos F4 no fueron inoculados, sino que se destinaron a producir y dar origen a semillas F5. Los lotes de semillas F5 se sembraron en 2017, y las plántulas derivadas fueron inoculadas con la misma cepa Dpc16 utilizada en la generación F3 (prueba de progenie). En el caso de las poblaciones PS-157 (P13 x P12) y PS-172 (P9 x P16), las semillas de las plantas F3 sobrevivientes de la prueba de progenie fueron igualmente sembradas en 2017 e inoculadas como plantas F4, con los datos obtenidos de esta generación se realizaron los cálculos para validar los resultados obtenidos en la F2 desde el enfoque de la genética cuantitativa.
Los números de individuos y familias evaluadas fueron los siguientes: 10 individuos de cada progenitor por año de evaluación; 74 familias F2:3 (740 individuos F3) y 66 familias F3:5 (660 individuos F5) para PS-258; 87 familias F2:3 (870 individuos F3) y 78 familias F4 (780 individuos F4) para PS-157; y 72 familias F2:3 (720 individuos F3) y 70 familias F4 (700 individuos F4) para PS-172.
Enfoque cualitativo
El análisis de segregación de los genes Rdc en cada generación estudiada se llevó a cabo realizando la prueba estadística no paramétrica de chi-cuadrado. De esta manera, se verificó la bondad de ajuste de las observaciones fenotípicas en familias F3 a una segregación genotípica 1:2:1 en la generación F2. Esta prueba también se aplicó para verificar la segregación individual en la generación F3 no agrupado en familias, cuya proporción esperada es una segregación fenotípica de 5 resistentes: 3 susceptibles.
Enfoque cuantitativo
Para el enfoque cuantitativo, en el análisis de valores medios se calcularon los grados de dominancia. En el análisis de varianzas, se estimaron la heredabilidad en sentido amplio y sentido estricto y el número de genes segregantes involucrados en cada cruzamiento. En cuanto a las covarianzas, se computaron las correlaciones fenotípicas, genotípicas y ambientales.
Determinación del grado de dominancia
Los valores genotípicos a, -a y d se calcularon de acuerdo con Kearsey y Pooni (2020) y con estos valores, se calcularon los grados de dominancia (d/a). Los valores genotípicos se calcularon como a = VFP1 - PO, -a = VFP2 - PO y d = VFF1 - PO. Tanto para incidencia como para severidad, VFP1 es el valor fenotípico del progenitor con mayor valor, VFP2 es el valor fenotípico del progenitor con menor valor, VFF1 es el valor fenotípico del híbrido F1, y PO (punto de origen) = (VFP1 + VFP2) /2.
Estimación de la heredabilidad de la resistencia a la CTS-Dpc
La heredabilidad en sentido amplio (H2) para incidencia y severidad en las generaciones F2 se estimó a partir del método de Mather, el cual estima la varianza ambiental a partir del promedio de las varianzas fenotípicas de padres y F1, posteriormente, resta la variancia fenotípica de la F2 a esta variancia ambiental para estimar la variancia genética y finalmente divide la varianza genética así calculada por la varianza fenotípica de la F2. En el caso del primer parámetro, se aplicó el método de componentes de varianza de acuerdo con las modificaciones propuestas por Cabodevila et al. (2017).
En las generaciones avanzadas, H2 para incidencia se estimó en cada cruzamiento utilizando la extensión del método de Mather propuesto por Kearsey y Pooni (2020), mientras que, para severidad, se aplicó un análisis de varianza (ANDEVA) a un criterio de clasificación para desglosar los componentes de varianza (Kearsey & Pooni, 2020).
Cuantificación del número de genes Rdc involucrados en la resistencia a CTS-Dpc
El número de genes (o loci, NG) Rdc segregantes involucrados en la resistencia CTS-Dpc se obtuvo aplicando la fórmula propuesta por Kearsey y Pooni (2020). La varianza aditiva se calculó relacionando el componente de varianza genética de la F2 con el componente entre familias correspondiente a cada generación avanzada bajo estudio. Se construyó un sistema de dos ecuaciones con dos incógnitas que presenta una única solución compatible. Estos cálculos asumen que los alelos son de efecto significativo y están en asociación, es decir, que el progenitor de mayor valor porta los alelos de cada locus estimado que aumenta los valores de los parámetros I % y S %, y el progenitor de menor valor, porta aquellos que contribuyen a los valores más bajos del parámetro para I % y S % (Kearsey & Pooni, 2020).
Para la evaluación de CTS-Dpc realizada, esto implica que el progenitor R tiene solo alelos que determinan menores valores de I % y S %. Del mismo modo, el progenitor susceptible porta solo alelos que aumentan estas variables. La varianza no aditiva se despejó conjuntamente con la varianza aditiva de los sistemas de dos ecuaciones con dos incógnitas planteados previamente (Kearsey & Pooni, 2020).
Cálculo de las correlaciones fenotípica, genotípica y ambiental entre la incidencia y severidad
La correlación genética entre I % y S % en las generaciones F2 se calculó despejando la covarianza genética como la diferencia entre la fenotípica y la ambiental. Posteriormente, la covarianza genética se dividió por el producto entre los desvíos genéticos calculados como la raíz cuadrada de las varianzas genéticas obtenidas en la estimación de H2. La covarianza ambiental fue despejada de la correlación entre incidencia y severidad para los progenitores, a través de la multiplicación entre el coeficiente de correlación r por el producto entre los desvíos para ambas variables.
El mismo procedimiento algebraico, pero con datos de F2, fue seguido para despejar la covarianza fenotípica. Para los cálculos de los coeficientes de correlación r se usó el programa InfoStat y se siguieron los pasos descritos por Kearsey y Pooni (2020). Las operaciones realizadas fueron: correlación fenotípica: rxy = correlación en F2. De aquí, se despeja la covarianza fenotípica como Cov. Fen.xy = rxy x desvío estándar x x desvío estándar y. Luego, se calculó la covarianza genética (Cov. Gen.xy) como la diferencia entre la covarianza fenotípica xy y la covarianza ambiental xy, esta última estimada como (Cov. P1 + Cov. P2 + Cov. F1) / 3, donde cada covarianza corresponde a rxy x desvío estándar x x desvío estándar y de la generación respectiva.
Finalmente, se obtuvo la correlación genéticaxy como rgxy = Cov. Gen.xy / (Var. Gen. x x Var. Gen. y)1/2. Estas varianzas genéticas fueron obtenidas previamente en el cálculo de H2. Las correlaciones fluctúan entre -1 y 1, valores que representan una completa asociación en sentido inverso o directo, respectivamente, entre las variables. Un valor igual a cero indica una falta de asociación entre ellas. En el caso de la partición de la correlación fenotípica en sus componentes genético y ambiental, y a diferencia de lo que ocurre con la heredabilidad, la suma de estos componentes no necesariamente reconstituye el total, ya que se trata de coeficientes y no exclusivamente de covarianzas.
Resultados
Enfoque cualitativo
Las proporciones fenotípicas (1:2:1) observadas en las pruebas de progenie de las familias F2:3 de las poblaciones PS-258 (GP13 x GP4), PS-157 (GP13 x GP12) y PS-172 (GP9 x GP16) permitieron inferir las proporciones genotípicas de los respectivos individuos progenitores en F2. A partir de estos datos, se realizaron los cálculos correspondientes a dicha generación, y estas poblaciones fueron evaluadas bajo un criterio de genética cualitativa. La población PS-258 (GP13 x GP4) mostró una segregación fenotípica 1:2:1 entre las familias F2:3 (prueba de progenie) y se pudo inferir el ajuste a una proporción genotípica mendeliana esperada de 1:2:1 (1RR: 2Rr: 1rr) y fenotípica de 3:1 (3R-: 1rr) en los individuos progenitores F2 (Cuadro 2).
Cuadro 2 Análisis cualitativo de la segregación del gen Rdc1 en la población PS-258 realizado en 2017 en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina.
| Generaciones evaluadas | N | Comportamiento | Razón esperada | Observado | χ2 c | ||
| R | Segreg. | S | |||||
| GP13 | 10 | R | - | 9 | 1 | ||
| GP4 | 10 | S | - | 2 | 8 | ||
| F2:3 | 74 | 1 R : 2 Segr : 1 S | 18,5 : 37: 18.5 | 21 | 42 | 11 | 4,05 ns |
| F3 | 740 | 5 R : 3 S | 466 | 274 | 0,07 ns | ||
N: número de plantas (o familias en F2:3) inoculadas / N: number of plants (or F2:3 families) that were inoculated
Observado: reacción de enfermedad (número de plantas o familias en F2:3 observadas como R: resistentes, Segreg.: segregantes y S: susceptibles) / Observed: disease reaction (number of plants or F2:3 families observed as R: resistant, Segreg.: segregant and S: susceptible).
ns: no existen diferencias significativas entre los valores observados y los esperados. / ns: no significant difference was observed between the expected and the observed values.
La población PS-157 (GP13 x GP12), que comparte el progenitor GP13 portador del gen Rdc1 de resistencia a CTS-Dpc, registró un ajuste fenotípico similar de 1:2:1 entre las familias F2:3 y el correspondiente ajuste genotípico 1:2:1 (1RR: 2Rr: 1rr) y fenotípico de 3:1 (3R-: 1rr) en los respectivos individuos progenitores F2, aunque solo fue posible luego de flexibilizar el número de plantas muertas-susceptibles (PMS) admitidas para un progenitor resistente (R). En este caso, se aceptaron dos PMS por cada diez plantas inoculadas (Cuadro 3) para las familias F2:3. Este criterio permitió establecer la existencia del gen de resistencia Rdc1 de carácter mendeliano y herencia monogénica dominante, en la PS-157 (Peruzzo et al., 2019). No obstante, no se logró ajustar la segregación mendeliana para plantas individuales en la F3 (5 R :1 S).
Cuadro 3 Análisis cualitativo de la segregación del gen Rdc1 en la población PS-157 realizado en 2017 en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina.
| Generaciones evaluadas | N | Comportamiento | Razón esperada | Observado | χ2 c | ||
| R | Segreg | S | |||||
| GP13 | 10 | R | - | 9 | 1 | ||
| GP12 | 10 | S | - | 3 | 3 | ||
| F2:3 | 87 | 1 R : 2 Segr : 1 S | 21,75 : 43,5: 21.75 | 14 | 48 | 25 | 3,75 ns |
| F3 | 870 | 5 R : 3 S | 439 | 435 | 53,81 ** | ||
N: número de plantas (o familias en F2:3) inoculadas. / N: number of plants (or F2:3 families) that were inoculated.
Observado: reacción de enfermedad (número de plantas o familias en F2:3 observadas como R; resistentes, Segreg.: segregantes y S: susceptibles). / Observed: disease reaction (number of plants or F2:3 families observed as R: resistant, Segreg.: segregant and S: susceptible)
ns: no existen diferencias significativas entre los valores observados y los esperados. / ns: no significant difference was observed between the expected and the observed values.
A diferencia de las poblaciones anteriores, ninguno de los progenitores de la población PS-172 (GP9 x GP16) porta el gen Rdc1, y sus generaciones segregantes no ajustaron a ninguna segregación mendeliana conocida. En la prueba de progenie F2:3 no se obtuvo una segregación fenotípica que permitiera inferir una segregación genotípica de 1:2:1 y fenotípica de 3:1 a CTS-Dpc en la población F2 de sus respectivos progenitores (Peruzzo, 2018). En consecuencia, no se presentan los análisis de bondad de ajuste, debido a la imposibilidad de contrastar los resultados con una segregación mendeliana conocida.
Enfoque cuantitativo
A partir de estos resultados experimentales obtenidos bajo criterio cualitativo, se inició el estudio de la herencia de la resistencia a CTS-Dpc desde un enfoque de la genética cuantitativa, el cual permitió ampliar y complementar los resultados alcanzados mediante un enfoque de la genética mendeliana clásica. La incidencia, expresada como porcentaje de plantas susceptibles y distribución fenotípica de la severidad, causadas por cancrosis de tallo en soja por Diaporthe phaseolorum var. caulivora (ciclo 2017) en cinco genotipos de soja seleccionados como progenitores en el plan de cruzamientos y mejoramiento frente a la enfermedad, mostraron una elevada variabilidad entre cultivares (Cuadros 4 y 5). Entre ellos, el progenitor GP13 es portador del gen Rdc1 de resistencia a CTS-Dpc, el GP9 porta dos genes Rdm de resistencia a CTS-Dpm, y los progenitores GP4, GP12 y GP16 corresponden a genotipos de interés por su valor agronómico.
Cuadro 4 Valores promedio de Incidencia y Severidad (%) de Cancrosis del Tallo (Diaporthe phaseolorum var. caulivora) registrados durante tres años, en cinco variedades de soja seleccionados como progenitores, desarrollados en el Campo Experimental J Villarino, Zavalla, Santa Fe, Argentina, en 2012, 2013 y 2017.
| Año | 2012 | 2013 | 2017 | Promedio 3 años |
| Progenitores | % Incidencia | |||
| GP13 | 9,1 | 0 | 20 | 9,7 |
| GP4 | 60 | 80 | 100 | 80 |
| GP12 | 60 | 40 | 60 | 53,3 |
| GP9 | 80 | 71,4 | 80 | 77,1 |
| GP16 | - | 50 | 40 | 45 |
| Progenitores | % Severidad | |||
| GP13 | 25 | 20 | 37 | 27,3 |
| GP4 | 72 | 86 | 73 | 77 |
| GP12 | 67,5 | 55 | 50,5 | 57,7 |
| GP9 | 85 | 77 | 74,5 | 78,8 |
| GP16 | - | 61 | 51 | 56 |
Cuadro 5 Distribución del valor fenotípico de la incidencia (I %) y la severidad (S %) de CTS-Dpc registrada en cinco progenitores de soja seleccionados y evaluados en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina, durante el ciclo 2017.
| Genotipo Progenitor | >0 hasta 30 Vr: 20 y 25 | >30 hasta 45 Vr: 45 | >45 hasta 60 Vr: 60 | >60 hasta 75 Vr: 75 | >75 hasta 100 Vr: 100 | Severidad % promedio | Incidencia % promedio |
| GP13 | 8 Vr: 6(20); 2(25) | 0 | 0 | 0 | 2 | 37,0 | 20 PMS |
| GP4 | 2 Vr: 30 | 0 | 2 | 2 | 4 | 73,0 | 80 PMS |
| GP12 | 4 Vr: 2(20); 2(25) | 1 | 2 | 2 | 1 | 50,5 | 80 PMS |
| GP9 | 2 Vr: 25 | 1 | 0 | 1 | 6 | 74,5 | 70 PMS |
| GP16 | 5 Vr: 4(20); 1(25) | 1 | 1q | 0 | 3 | 51,0 | 50 PMS |
PMS = Plantas muertas-susceptibles. / PMS = Dead Ð susceptible plants.
En cada celda se indica el número total de individuos (en mayor tamaño) y el desglose de ese número total (en menor tamaño) en función de su valor registrado (Vr) para cada categoría o rango de severidad en porcentaje (S%) según escala del Cuadro 2, previo a las columnas de los promedios de S% y valor de Incidencia (I%), en el ciclo 2017. / In each cell the total number of individuals is indicated (in larger size) and the breakdown of that total number (in a smaller size) based on their registered Value (Vr) for each category or severity range in percentage (S%) according to the scale in Table 2, prior to the columns of the averages of S% and incidence value (I%), in the 2017 cycle.
Los resultados correspondientes a los parámetros genéticos se presentan por población, con el fin de facilitar la interpretación de las implicaciones biogenéticas y agronómicas de tales estimaciones. A partir de los valores de severidad (S %) de los progenitores y su respectiva F1 en cada una de las tres poblaciones en estudio, se calculó de acuerdo con lo indicado en materiales y métodos, el valor PO, el valor genotípico a para ambos progenitores y el valor genotípico d, que los relaciona con la F1, así como el grado de dominancia para el parámetro de S %. Para incidencia (I %), al no contar con un valor para la F1 por falta de repeticiones, no fue posible calcular ni el valor de d ni el cociente d/a.
En todos los casos, los datos de las generaciones tempranas (F2:3) son los ya presentados para el enfoque cualitativo y corresponden a experimentos realizados en 2017. El enfoque de este nuevo análisis se hizo según criterios de la Genética Cuantitativa clásica. Los datos de las generaciones avanzadas (F3:5 para PS-258 y F4 para PS-157 y PS-172) provienen de experimentos realizados en 2020 y solo se analizaron mediante un enfoque cuantitativo.
Cruzamiento GP13 x GP4 - PS-258
En las Figuras 1 y 2 se muestran los valores genotípicos (a y PO) para la incidencia (I %) y la severidad (S %) correspondientes a los progenitores GP13 x GP4. En cuanto a heredabilidad y al número de genes, calculadas en las generaciones segregantes, en la generación F2:3 (n = 74 plantas) la media de I % fue de 57,43 ± 21,84, con una heredabilidad de 0,68. En la generación F3:5 (n = 17 familias) la media fue 9,62 ± 25,56. En este caso, no se detectaron diferencias significativas entre familias, por lo que la heredabilidad, aunque presentó un valor de 0,17, no resultó significativamente distinta de cero. En consecuencia, no fue posible calcular el número de genes involucrados en esta población.
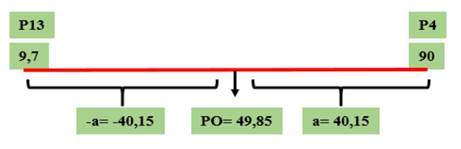
Figura 1 Valor genotípico a y PO para el parámetro de incidencia (Cuadro 3) correspondientes al cruzamiento GP13 × GP4, evaluado en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina, durante los ciclos 2012, 2013 y 2017.
El cálculo del grado de dominancia para el parámetro de S % indicó un valor de -1,72, lo cual evidencia acciones de sobredominancia orientadas hacia valores menores, correspondientes al progenitor portador del gen Rdc1. En consecuencia, se observa heterosis, ya que, como se muestra en la Figura 3, la F1 supera el valor del progenitor con menor S %. Este comportamiento aporta relevancia agronómica, dado que la F1 presenta una mayor resistencia que el progenitor portador de Rdc1.
En cuanto a la heredabilidad y el número de genes, en la generación temprana F2:3 la media de S % fue de 53,68 ± 15,60, con una heredabilidad de 0,55, mientras que en la generación avanzada F3:5 la media S % fue 9,96 ± 30,01. Las diferencias entre familias fueron estadísticamente significativas, lo que permitió calcular una heredabilidad significativa de 0,18. Asimismo, al combinar las varianzas aditivas significativas en las generaciones tempranas y avanzadas, fue posible calcular el número de genes, que resultó igual a 0,78 (aproximado a 1).
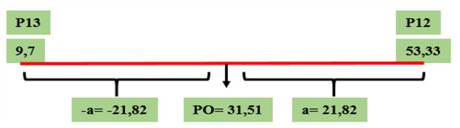
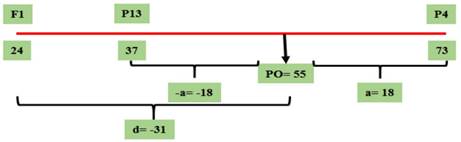
Figura 3 Valor genotípico a y PO para el parámetro de incidencia correspondientes al cruzamiento GP13 × GP12, evaluado en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina, durante los ciclos 2012, 2013 y 2017.
De acuerdo con estos valores, en la PS-258 derivada de GP13 x GP4, para el parámetro de severidad, tanto la media como la heredabilidad disminuyeron desde las generaciones segregantes tempranas (F2:3) hacia las tardías (F3:5). Esto puede deberse al efecto de la selección sobre un único locus Rdc1 para el fenotipo dominante que otorga resistencia, en congruencia con el grado de dominancia y el número de genes estimado. Finalmente, en este cruzamiento la correlación fenotípica entre incidencia y severidad fue de 0,88, en tanto que la genética y la ambiental fueron 0,89 y 0,96, respectivamente.
Dado que la correlación integra el comportamiento conjunto de I % y S %, los valores elevados y positivos de las tres correlaciones son congruentes con el comportamiento similar de las medias y las varianzas de ambos parámetros desde las generaciones segregantes tempranas hacia las tardías: una reducción en la media y un aumento en las varianzas fenotípicas. En este contexto, la reducción de la media se atribuyó a la selección de plantas resistentes. El aumento en la varianza puede deberse a la segregación del locus Rdc1, ya que la selección de los fenotipos más resistentes implica una selección de heterocigotas, de acuerdo con el valor de d/a.
Cruzamiento GP13 x GP12 - PS-157
Los valores genotípicos para I % y S %, respectivamente, en este cruzamiento se muestran en las Figuras 3 y 4. En cuanto a heredabilidad y el número de genes, en la generación temprana F2:3 (n = 87 plantas) la media de I % fue de 49,54 ± 22,20, con una heredabilidad de 0,76, mientras que en la generación avanzada F4 (n = 59 familias) la media fue 9,54 ± 15,01. Las diferencias entre familias fueron estadísticamente significativas, lo que permitió calcular una heredabilidad significativa de 0,48; asimismo, el número de genes resultó igual a 4,1.
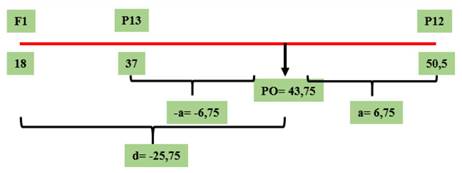
Figura 4 Valores genotípicos (a y d) y PO calculados para el parámetro de severidad correspondientes al cruzamiento GP13 × GP12, evaluado en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina, durante los ciclos 2012, 2013 y 2017.
En este cruzamiento, al igual que en el primero, el grado de dominancia presentó el mismo sentido que el cruzamiento GP13 x GP4, con un valor de -3,81, lo que indica acciones de sobredominancia orientadas hacia los menores valores. La F1 resultó más resistente que el progenitor portador del gen Rdc1. En cuanto a heredabilidad en sentido amplio y al número de genes, en la generación F2:3 la media de S % fue de 55,94 ± 16,80, con una heredabilidad en sentido amplio de 0,60; mientras que en la generación avanzada F4 la media de S % fue de 23,35 ± 13,65, observándose diferencias significativas entre las familias.
La heredabilidad en sentido estricto fue, en consecuencia, significativa, con un valor de 0,21. Al combinar las varianzas aditivas significativas en las generaciones tempranas y avanzadas, el número de genes aportados por el progenitor resistente resultó igual a 0,18 (aproximado a 0). Esto permitiría inferir, de acuerdo con lo definido en materiales y métodos, que el modelo matemático considera que los alelos de cada gen que aumentan o disminuyen la media de un carácter están en asociación, es decir, son aportados por un único progenitor, que en este cruzamiento tanto el progenitor resistente como el susceptible aportan alelos de resistencia en los distintos loci, tal como fue calculado para I %. Sin embargo, para S % el balance neto en las generaciones segregantes resultó nulo.
En este cruzamiento GP13 x GP12 la correlación fenotípica entre incidencia y severidad fue de 0,96, mientras que la genética y la ambiental fueron 1,00 y 0,91, respectivamente. Dado que las correlaciones integran el comportamiento de ambos parámetros para este cruzamiento GP13 x GP12, se puede observar que, tanto para incidencia como para severidad, y al igual que en el cruzamiento anterior (GP13 x GP4), la media y la varianza presentan comportamientos similares desde las generaciones segregantes tempranas hacia las avanzadas. No obstante, en este cruzamiento ambas medidas estadísticas disminuyeron.
En cuanto a la media, al igual que el cruzamiento GP13 x GP4, la reducción se atribuye al efecto de la respuesta a la selección de plantas resistentes. En cuanto a la varianza, la diferencia puede deberse a que en el cruzamiento anterior la selección operaba sobre un único locus Rdc1. En este caso, y de acuerdo con el número de genes estimado en el párrafo anterior, la reducción obedecería a que la selección está operando no solo sobre Rdc1 aportado por GP13, sino por otros loci Rdc aportados por GP12, cuya identificación requeriría la realización de cruzamientos adicionales.
Cruzamiento GP9 x GP16 - PS-172
Los valores genotípicos para I % y S %, respectivamente, en este cruzamiento se muestran en las Figuras 5 y 6. En cuanto a heredabilidad y número de genes, en la generación temprana F2:3 (n = 64 plantas) la media de I % fue de 59,69 ± 18,85, con una heredabilidad de 0,89, mientras que en la generación avanzada F4 (n = 58 familias) la I % media fue 46,23 ± 22,63. Las diferencias entre familias fueron estadísticamente significativas.
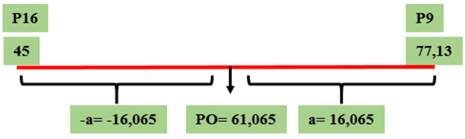
Figura 5 Valor genotípico a y PO para el parámetro de incidencia correspondientes al cruzamiento GP9 × GP16, evaluado en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina, durante los ciclos 2012, 2013 y 2017.
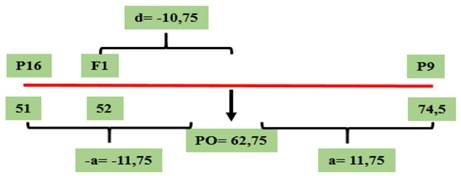
Figura 6 Valores genotípicos (a y d) y PO calculados para el parámetro de severidad correspondientes al cruzamiento GP9 × GP16, evaluado en el Campo Experimental J. Villarino, Zavalla, Santa Fe, Argentina, durante los ciclos 2012, 2013 y 2017.
La heredabilidad resultó significativa, con un valor de 0,92 y el número de genes con efecto significativo aportado por el progenitor más resistente fue igual a 0,2, que se redondea a 0. Esta observación es consistente con el hecho de que ninguno de estos progenitores es portador de genes mayores conocidos de resistencia a la CTS-Dpc y, por lo tanto, el aumento de la heredabilidad a lo largo de las generaciones de autofecundación (evidenciado en la disminución de la incidencia en F4 al comparar con F2:3) se atribuiría a la acumulación de poligenes.
En este cruzamiento, en el cual los progenitores no poseen el gen Rdc1, el grado de dominancia también presentó sentido negativo (-0,91), lo que indica dominancia parcial, como puede observarse en la Figura 6. Si bien la F1 presenta un valor cercano al progenitor con menor valor, es clasificada como susceptible de acuerdo con la escala utilizada para la evaluación de la enfermedad.
En cuanto a la heredabilidad y al número de genes, en la generación temprana F2:3 la media de S % fue de 66,84 ± 15,07, con una heredabilidad de 0,04, mientras que en la generación avanzada F4 la S % media fue 55,51 ± 18,78. Las diferencias entre familias fueron significativas, lo que permitió calcular una heredabilidad significativa de 0,13. Asimismo, al combinar las varianzas aditivas significativas en las generaciones tempranas y avanzadas, fue posible calcular el número de genes, que resultó igual a 0,24 (redondeado a 0). Las observaciones realizadas para incidencia, en cuanto a que el número estimado de genes de efecto significativo es nulo y coincide con la ausencia del gen Rdc1 en ambos progenitores, son igualmente válidas para severidad.
En este cruzamiento GP9 x GP16 la correlación fenotípica entre I % y S % fue de 0,99, mientras que la correlación genética también fue de 0,99. La correlación ambiental, si bien tuvo un valor de 0,89, no resultó estadísticamente significativa. La integración del comportamiento conjunto de ambas variables en estas correlaciones indica que, en este cruzamiento, en el que no participa el progenitor GP13 portador del gen Rdc1, se observa un compartimiento diferenciado respecto de los cruzamientos anteriores.
Las medias de incidencia y severidad, tanto en la generación segregante temprana F2:3 como en la generación avanzada F4, presentaron valores más elevados que los observados en las generaciones correspondientes de los otros dos cruzamientos, debido a la ausencia del gen de resistencia Rdc1. Además, en este cruzamiento, la reducción de las medias desde la generación temprana hacia la avanzada, no fue tan marcada como en los cruzamientos anteriores, aun cuando también se seleccionaron plantas resistentes. Este comportamiento es congruente con la presencia de poligenes de efecto menor asociados a la resistencia, en lugar de genes de efecto significativo, detectados previamente.
En cuanto a la varianza fenotípica, se observa que este cruzamiento mostró un comportamiento similar al del cruzamiento GP13 x GP4, ya que en la generación F4 fue mayor que en la F2:3, en coincidencia con la presencia de más de un gen involucrado. Sin embargo, en el cruzamiento GP9 x GP16 las heredabilidades aumentaron de las generaciones segregantes tempranas a las avanzadas, a diferencia de la disminución observada en los otros dos cruzamientos. Este resultado coincide con una mayor varianza fenotípica y genética, causada por la segregación y recombinación de poligenes de resistencia durante los ciclos de autofecundación.
Cabe destacar que, si bien en el cruzamiento GP13 x GP4 también se detectó la participación de más de un gen, al menos para el parámetro de incidencia, su efecto fue mayor, correspondiente a un gen de herencia mendeliana. En GP9 x GP16, en cambio, la base genética de la resistencia se asocia a poligenes de efecto menor. En este cruzamiento, las correlaciones también mostraron un comportamiento diferencial respecto de los cruzamientos anteriores, dado que la correlación ambiental resultó no significativa.
Discusión
Uno de los métodos de control de las enfermedades es, sin duda, la resistencia genética. Este método es considerado el más eficaz, económico y ambiental sustentable, ya que no implica contaminación del ambiente. Sin embargo, esto no significa la exclusión de otros métodos, entre ellos el empleo de fitosanitarios o el manejo cultural (Oliveira Nogueira et al., 2015).
Desde la genética cualitativa, solo se obtuvo un ajuste satisfactorio en una de las poblaciones analizadas (PS-258). En las otras dos poblaciones evaluadas, el ajuste se logró, como se indicó en resultados, mediante la flexibilización de criterios para considerar “planta resistente” (PS-157) o no se logró ningún ajuste (PS-172). Por este motivo, se consideró oportuno aplicar un enfoque de genética cuantitativa, debido a que pueden existir genes menores y modificadores, adicionalmente al gen mayor Rdc1, reportado por Peruzzo et al. (2019).
Desde la genética cuantitativa, el cálculo de la heredabilidad en sentido amplio (H2) para las poblaciones en estudio mostró valores altamente variables entre poblaciones y generaciones. Para el cruzamiento particular GP13 x GP4, la selección resultó en plantas fenotípicamente más homogéneas, es decir, más similares en sus valores de mejora o breeding values, para la resistencia (Bayer et al., 2022), lo que condujo a una disminución de la varianza genética aditiva para el carácter severidad, y en consecuencia, de la heredabilidad en sentido estricto (Kearsey & Pooni, 2020). Esto es congruente con la disminución observada en la media de severidad en la generación segregante avanzada.
Una selección efectiva contribuye al desplazamiento de la media de la población, por lo cual es de esperarse el agotamiento progresivo de la varianza aditiva y, por ende, la disminución de la heredabilidad (Cabodevila et al., 2017). Si bien en este cruzamiento se observa un aumento de la varianza en la generación segregante avanzada, es necesario considerar que dicha generación corresponde a una F3:5. Esto implica una alta segregación intrafamiliar, debido a que la selección en F3, combinada con una alta segregación interfamiliar, produjo líneas homocigotas discrepantes en F5.
En estudios mendelianos en este cruzamiento fue necesario flexibilizar el criterio de clasificación (de 1 PM a un máximo de 2 PM de 10 plantas evaluadas) para caracterizar el comportamiento fenotípico de las familias F2:3. De esta manera, fue posible inferir el genotipo de los progenitores de la F2 y lograr un ajuste a la proporción mendeliana genotípica 1:2:1 y fenotípica 3:1, esperadas para dicha filial. Esta modificación se realizó con base en el criterio de la escala de severidad presentada en el Cuadro 1, la cual asignó un valor a cada uno de los individuos segregantes F3 distribuidos en familias F2:3 y fue empleada para categorizar como genotipos estables o cultivares resistentes a CTS a aquellos con ≤ 25 % de plantas muertas (PM) (Chiesa et al., 2013; López Achaval et al., 2014).
En este trabajo se aplicó a poblaciones segregantes tempranas el criterio de que las familias F2:3 que registraran el umbral de menos de 2 PM sobre 10 plantas inoculadas fueran consideradas provenientes de un progenitor RR. La necesidad de flexibilizar el criterio de evaluación para ajustar a la segregación esperada podría deberse a la presencia de genes modificadores que no estarían en asociación, es decir, no presentes únicamente en un solo progenitor (GP13, el más resistente), sino en dispersión (Kearsey & Pooni, 2020). En otras palabras, la resistencia en este segundo cruzamiento estaría determinada por diferentes loci presentes tanto en el progenitor GP13 como en el GP12, de manera similar a lo reportado en otras interacciones del complejo Diaporthe phomopsis - soja (Hernández, 2022).
En este contexto podría inferirse que ambos progenitores, el más resistente y el más susceptible, aportan genes de resistencia (R). En concordancia con ello, puede explicarse que la F1 presente un patrón de sobredominancia hacia valores menores, más pronunciado en comparación con el cruzamiento anterior. Si bien la presencia de genes de resistencia en ambos progenitores puede otorgar a la planta una mayor protección frente a la CTS-Dpc, debe considerarse que la fijación de estos genes resulta más compleja en comparación con la herencia controlada por un gen mayor único.
Para el parámetro de I %, también en este cruzamiento (GP13 x GP12) se verificó la tendencia a comportarse como un carácter cualitativo, debido a la forma de medición utilizada. En consecuencia, es esperable encontrar un mayor valor de H2 en comparación al parámetro de S %. La disminución del valor de H2 observada en la generación segregante avanzada respecto de la temprana concuerda con la respuesta a la selección, dado que esta población se formó a partir de las plantas R que sobrevivieron en la generación anterior.
En el cruzamiento GP9 x GP16, los valores de H2 mostraron un comportamiento diferente comparado con las otras dos poblaciones (GP13 x GP4 y GP13 x GP12). Esto fue especialmente notable para el parámetro I %, que presentó un valor más elevado en la generación F4 que en la generación F2:3, cuando lo esperable sería una disminución de la heredabilidad a medida que avanzan las generaciones. No obstante, si bien el P9 cuenta con genes de resistencia a CTS-Dpm, no sería portador de genes de resistencia (Rdc) a CTS-Dpc identificados, fundamentalmente frente al aislamiento Dpc16, utilizado en el presente trabajo.
En consecuencia, es de esperarse una población con mayor susceptibilidad en comparación con las otras dos poblaciones. Sin embargo, es oportuno señalar que, en la evaluación de interacciones entre dieciséis cultivares de soja y cuatro aislamientos de Dpc causales de CTS (interacción planta-patógeno, IPP), se detectaron IPP específicas que muestran al GP9 y GP16 con comportamientos significativamente disímiles; es decir, reaccionaron como susceptibles (S) o resistentes (R) frente a diferentes cepas Dpc evaluadas. Estas respuestas específicas permiten inferir la posible presencia de otros genes Rdc, diferentes al Rdc1, en los progenitores GP9 y GP16.
Si bien la media de la generación segregante avanzada presentó un descenso, dicha disminución fue considerablemente menor que la observada en las otras dos poblaciones. Los valores elevados de H2 observados para I % podrían explicarse por el hecho de que la R expresada sea horizontal basal, compuesta por un sistema oligogénico o poligénico, y no exclusivamente atribuible a un gen mayor único. Con el avance de los ciclos de autofecundación, se manifiestan diferentes mecanismos de R, lo que explicaría el mayor valor de H2 en la generación segregante avanzada, sin dejar de considerar que la medición de este parámetro no se realiza como un carácter cuantitativo ordinal, sino con un comportamiento más cercano al cualitativo.
Valores similares de heredabilidad en sentido amplio para el parámetro de S % fueron descritos por Sulistyo y Sumartini (2016) para la S % de la roya asiática (Phakopsora pachyrhizi) en soja (0,22; 0,18 y 0,14), una, dos y tres semanas después de la inoculación, respectivamente. En el presente trabajo, las poblaciones que compartieron el progenitor resistente GP13, portador del gen Rdc1, presentaron valores de heredabilidad medios a altos. En la PS-172, ante la ausencia de dicho gen, la variación fenotípica observada se atribuiría a factores externos y a efectos genéticos menores (poligenes).
La correlación genética (CG) es crucial en el mejoramiento pues representa el grado de asociación causada por ligamiento genético o pleiotropía entre dos caracteres cuantitativos (Cabodevila et al., 2017). Desde este punto de vista, puede presentar tanto efectos positivos como negativos, dado que permite la selección indirecta de un carácter “Y”, de difícil medición por la complejidad o por la imposibilidad de evaluarlo en etapas tempranas, a través de un carácter “X”, de medición más sencilla. Sin embargo, este beneficio puede verse contrarrestado por el hecho de que una correlación genética existente provoque una selección no deseada o “arrastre por ligamiento”, debido a una asociación agronómicamente desfavorable entre dos caracteres.
En este trabajo, la correlación genética (CG) para los cruzamientos que comparten el padre resistente (GP13) presentó valores altos y significativos, de 0,89 y 1,00 para los cruzamientos GP13 x GP4 y GP13 x GP12, respectivamente. Si bien ambas poblaciones presentan correlaciones positivas, ésta se acentuó más en la población que mostró el valor de H2 más elevado, la cual podría contar con genes en dispersión (Kearsey & Pooni, 2020), ubicados en diferentes loci aportados por ambos progenitores (GP13 x GP12). Esta interpretación podría verse respaldada por lo observado previamente, dado que GP12 reaccionó como genotipo S frente a dos cepas Dpc y como moderadamente resistente/moderadamente susceptible (MR/MS) frente a otras dos cepas diferentes de Dpc.
Respecto al cruzamiento GP9 x GP16, la correlación ambiental resultó no significativa debido a que en los padres no se observó asociación entre S % e I %. En este caso, ante la ausencia del gen Rdc1, las respuestas de las plantas podrían corresponder a ajustes estructurales o fisiológicos, sin una base genética claramente definida. Además, al no existir correlación ambiental, las correlaciones fenotípica y genética deben asumirse como equivalentes y, aunque resultaron elevadas (0,99), no deberían considerarse criterios confiables para su utilización directa en un programa de mejoramiento genético.
Contrario a lo observado en este trabajo, correlaciones genéticas superiores a las correlaciones fenotípica y ambiental fueron informadas en una investigación sobre las variables que influyen en mayor medida sobre la calidad del fruto y el rendimiento del tomate de árbol, con el fin de ser utilizadas como criterios de selección (Lagos Santander et al., 2013). Solo en el cruzamiento GP13 x GP12, la correlación genética presentó un comportamiento similar al presentado por Lagos Santander et al. (2013). Estas correlaciones podrían resultar útiles en un programa de mejoramiento, ya que facilitan la toma de decisiones al momento de seleccionar individuos resistentes utilizando S % o I %, parámetros que simplifican el proceso de evaluación al ser medidos de forma cualitativa, lo que permitiría ahorrar tiempo y esfuerzo.
En este trabajo se validó la presencia del locus Rdc1 de resistencia a CTS-Dpc en el progenitor GP13 mediante un estudio cuantitativo. Para ello, se evaluaron dos poblaciones que compartían el progenitor resistente GP13, con dos progenitores susceptibles (GP13 x GP4; GP13 x GP12), así como una tercera población obtenida a partir del cruzamiento entre GP9 x GP16. En ésta, GP9 porta dos genes Rdm efectivos para CTS-Dpm.
El número de genes estimados para el cruzamiento GP13 x GP4, para el parámetro de severidad fue de 0,78, valor que, en términos prácticos y desde el punto de vista biológico, se redondea a uno. Este resultado coincide con lo identificado mediante enfoques mendelianos clásicos y con la teoría gen a gen de Flor, la cual postula que los alelos de resistencia (R) dominan sobre los alelos susceptibles (r), y con el grado de dominancia, que indica acciones de sobredominancia orientadas hacia valores menores de severidad o enfermedad, es decir, hacia los fenotipos resistentes, de interés agronómico (Burbano-Figueroa, 2020). Para el parámetro I %, el número de genes no pudo ser estimado debido a la no significancia de la varianza aditiva.
En el cruzamiento GP13 x GP12 para el parámetro de S % el número de genes fue de 0. La ausencia de genes mayores no implica su inexistencia, sino que podría deberse a que tanto el progenitor GP13 como GP12 aportan genes en dispersión, localizados en diferentes loci (Kearsey & Pooni, 2020). Esto puede observarse en la Figura 6 donde se aprecia un mayor grado de sobredominancia en la F1 en comparación con el cruzamiento anterior, fenómeno que podría atribuirse a la acumulación de genes aportados tanto por GP13 como, posiblemente, por GP12.
El número de genes encontrados para el parámetro I % fue de 4. Esta discrepancia entre severidad e incidencia en cuanto al número de genes podría explicarse por el hecho de que, para I %, los genes R se encuentran en asociación, en contraste con lo observado para el parámetro de S %. En esta población fue necesario flexibilizar los criterios para definir plantas PSR y familias F2:3, a fin de lograr el ajuste a la segregación fenotípica 1:2:1 (en prueba de progenie), correspondiente a la frecuencia genotípica de los progenitores en F2, cuya frecuencia fenotípica esperada es 3:1 (3R : 1S). Esta situación podría haber contribuido a la discrepancia observada.
En el cruzamiento GP9 x GP16, el progenitor GP9 porta dos genes de resistencia (Rdm4-5), carga genética altamente efectiva frente a CTS-Dpm (Chiesa et al., 2017). Sin embargo, este gen no confiere resistencia a la CTS causada por los tres aislamientos de Dpc evaluados en trabajos previos (Pioli et al., 2019). Estos resultados respaldan los obtenidos mediante enfoques mendelianos clásicos, que identificaron a GP13 como portador del gen Rdc1, y reafirman la ineficiencia del gen Rdm frente a CTS-Dpc.
Si bien el gen Rdc1 ya ha sido validado en el progenitor GP13, resulta necesario continuar con la búsqueda de nuevas fuentes de resistencia, considerando la variabilidad potencial del patógeno a lo largo del tiempo. En ese sentido, se vuelve imprescindible complementar los estudios de R vertical con enfoques cuantitativos, ya que estos permitirían identificar genes modificadores, así como profundizar la evaluación del comportamiento del cruzamiento GP9 x GP16 desde un enfoque cuantitativo, con el objetivo de garantizar la estabilización de la resistencia observada durante los distintos ciclos de autofecundación frente a CTS-Dpc.
Conclusiones
El análisis del cruzamiento GP13 x GP12 (PS-157) y el mayor grado de sobredominancia observado en la F1 ponen de manifiesto que, en la resistencia a CTS-Dpc, podrían estar involucrados no solo genes mayores en asociación, sino también genes en dispersión localizados en diferentes loci. Los resultados obtenidos mediante la aplicación integrada de enfoques cualitativos y cuantitativos, utilizados como herramientas complementarias, permitieron revalidar al gen Rdc1, portado por GP13, como una fuente efectiva de resistencia genética de la soja frente a CTS-Dpc.

