











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
Permalink
The rabies virus infects different mammalian species worldwide (Sadeuh-Mba et al., 2017). Infection occurs due to the introduction of saliva from an infected animal through bites, wounds, or scratches on mucous membranes of animals (Bano et al., 2016).
The incubation period varies from 10 days to some years depending on the strain, extent, depth of the wound, the distance of the inoculation place, the particularities of the central nervous system, and inoculated doses (Salomão et al., 2017). After symptoms appear, a mortality rate of 100% occurs, which justifies its importance for public health and the economic losses for livestock farmers (Amarilla et al., 2018; Johnson et al., 2014).
The transmission occurs in two cycles based on the viral lineage of the host-vector. The terrestrial cycle is caused by carnivorous mainly transmitted by dogs (urban rabies), and bats mediate the aerial cycle, being the D. rotundus the primary source in America (Brito-Hoyos et al., 2013; Yaguana & López, 2017).
In Costa Rica, the Animal Health Service of Costa Rica (SENASA) and the Ministry of Agriculture and Livestock monitored rabies cases closely after implementing surveillance programs in 1985 (Hutter et al., 2016; León et al., 2021). However, infection cases by bats still happen in wild animals, livestock, and humans (Badilla et al., 2003; Hutter et al., 2016)
In Costa Rica, several researchers conducted rabies virus studies. Badilla et al. (2003) analyzed the possibility of rabies reemergence in humans following two cases presented in the country in 2001 after 31 years of absence (Badilla et al., 2003). Hutter et al. (2016) conducted a comprehensive epidemiological analysis of rabies in Costa Rica between 1985 and 2014 by analyzing outbreaks reported in cattle using nucleoprotein sequences. Another study (2018) analyzed how assessing changing weather and the El Niño Southern Oscillation impacts cattle rabies outbreaks and mortality in Costa Rica (Hutter et al., 2016). Streicker et al. (2019) conducted a nucleoprotein sequence-based phylodynamic study of the extinction-recolonization dynamics of the virus (Streicker et al., 2019), and León et al. (2021) made a resume of the historical occurrence of canine rabies and its elimination in Costa Rica. Also, discuss the ecology of the vampire bat as a reservoir and its management. As well, details the clinical characteristics of recent cases of human rabies (León et al., 2021).
Phylogenetic analyses are helpful in epidemiology, diagnostics, phylogeographic, evolutionary studies, and virus taxonomy. These provide an evolutionary perspective on the variation of traits determined for a group of viruses (Gorbalenya & Lauber, 2017). Phylogenetic studies of rabies are significant to understand the dynamics of virus spread. In the past, Streicker et al. (2019) observed that in Costa Rica, rabies outbreaks seem to originate from North America and South America. These are stationary and become extinct in the Costa Rican territory before crossing a neighboring country (Streicker et al., 2019). These phylogenetic studies are based on partial or complete genes like nucleocapsid or glycoprotein (Garcés-Ayala et al., 2017; Kuzmin et al., 2012; Streicker et al., 2019). However, no studies so far have drawn phylogenetic inferences using complete genomes compared with partial sequences. Advances in high-throughput nucleic acid sequencing technologies and bioinformatics methods have revolutionized phylogenetic analysis. These methods brought new perspectives for the research of viral infections (Drummond et al., 2002, 2012).
Assemblies of complete genomes with proper depth of coverage allow exploration overtime of the evolution of a viral genome during the infection process (Maurier et al., 2019) and to study possible mutations related to pathogenesis. Also, phylogenetic analysis of different regions of the genome shows some differences due to the recombination process. Currently, there are no whole-genome phylogenetic studies for rabies virus derived from Costa Rica isolates. This study conducts phylogenetic analyses using the complete genomes of recent isolates and compares the outcome to the one obtained using nucleoprotein gene studies.
MATERIALS AND METHODS
Samples: Four bovine brain tissue samples from different parts of the country were used in this study (Fig. 1). Tissue samples were collected during 2018 and tested positive for rabies by direct immunofluorescence and end-point PCR, as routine diagnostic done by the National Veterinary Services Laboratory (LANASEVE), SENASA. Pools of the cortex, hippocampus, thalamus, or medulla fragments were extracted for each sample and labeled as D01-18 (MW506939), D02-18 (MW506938), D03-18 (MW506937), and D04-18 (MW506940). Numbers assigned in the GenBank database are in parentheses. We conserved aliquots of tissue samples and extractions at -80C° and used nuclease-free vials and reagents to prevent RNA degradation.
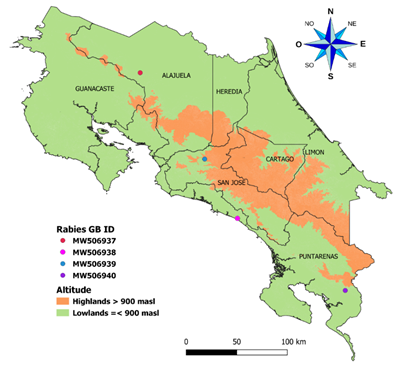
Fig. 1 The geographic distribution of genomes obtained in this study was identified in the region of Costa Rica. The map shows the political division of Costa Rica. One sample was taken at more than 900 meters above sea level and the other three at less than 900 meters above sea level. The samples were taken throughout the country.
RNA extractions and processing: To obtain the genomes for the four samples isolated, we performed total RNA extraction using the phenol-chloroform method, and then silica columns (Phasemaker™ Tubes) were used. We remove the ribosomal RNA portion using Exonuclease Terminator™ 5'- Phosphate-Dependent (Lucigen), then we treated it with DNAse I (Ambion) and cleaned it with RNAClean XP magnetic beads (Beckman Coulter). We determined post-processing RNA quality values by NanoDrop (Desjardins & Conklin, 2010). Since some regions sequences are complex to sequencing and can lead to low or no coverage, we designed virus-specific primers and combined them with previously published primers (Marston et al., 2013) and random hexamers to enhance the generation of ssDNA from viral RNA templates. Table 1 shows each primer's binding sites based on the reference genome (AB519641.1). We selected primer pairs according to favorable thermodynamic conditions and calculated the entropy values using Bio Edit v7.2 software (Hall et al., 2011) in the consensus regions.
We generated single-stranded DNA (ssDNA) with the Superscript III kit (ThermoFisher) using Random Hexamers (25pmol) and equimolar pooling (2µM) of the primers designed for this study (Table 1) as well as published primers (Marston et al., 2013). RNAse H (Sigma Aldrich) and Klenow enzyme (Promega) were added to the pre-reaction as previously described (Moser et al., 2016) to generate double-stranded DNA (dsDNA) for each sample. The obtained dsDNA was cleaned with AMPure XP magnetic beads (Beckman Coulter) and quantified following the QuantiFluor dsDNA System protocol (Promega).
TABLE 1 Rabies-specific designed primers used for ssDNA generation
| Identification | Sequence 5'- '3 | Genome binding site 1 |
|---|---|---|
| PFowCG1 | ACGCTTAACAACAAAATCAGAGAAGAAGYAG | 1 - 31 |
| PRevCG1 | TGACACCTCTCTCTGACTTTRAAACAC | 4 401 - 4 427 |
| PFowCG2 | CAACAAGACYTTGATGGAGGCTGA | 4 326 - 4 349 |
| PRevCG2 | GAGACAGGATCWGAGAACTGACGTATG | 8 138 -8 163 |
| PFowCG3 | GTCAACCTTGCCAATATAATGTCAA | 7 869 - 7 893 |
| PRevCG3 | TGAGCAGAGCAGACCCARTCT | 10 307 - 10 327 |
| PSRevCG1.2 | CTGGCACATTTTRCGACTCCTTG | 2 227 - 2 249 |
| PSFowCG2.2 | GACTTAGTCTTTGTGTAYGGCTGTTACAG | 6 486- 6 514 |
1Primer binding positions are based on the reference genome AB519641.1
Library Preparation and NGS sequencing: We prepared four genomic libraries (one for each isolate) following the Nextera XT protocol (Illumina) according to the manufacturer's instructions with minor variations. Initial starting dsDNA for tagmentation was used at a concentration of 0,3ng/µl and then amplified. Individual libraries were equimolar pooled (2nM) and denatured following Illumina standard normalization protocol A. These were sequenced on an Illumina MiSeq platform using a paired-end (2x250-bp) protocol. The four libraries obtained was sequenced.
Bioinformatic analyses: Genomes were assembled and processed using the Genome Detective tool (Vilsker et al., 2019) with the default settings, which include a pipeline with Trimmomatic (Bolger et al., 2014), FastQC (Brown et al., 2017), DIAMOND (Buchfink et al., 2014), metaSPADES (Bankevich et al., 2012), and Advanced Genome Aligner (AGA) (Deforche, 2017). Annotation was done with Geneious Prime 2020.2.4 software and verified by Blast analysis (NCBI Resource Coordinators, 2018). Genomes were aligned with ClustalW (Thompson et al., 1994) to observe the identity percentage.
Phylogenetic analysis: We assessed the potential recombination events among the complete rabies genomes using the RDP4 v.4.39 software (Martin et al., 2015) to prevent bias in evolutionary inferences. Newly assembled genomes were aligned with MAFFT V.7.427 (L-INS-i mode) (Katoh & Standley, 2013) on the CIPRES server (Miller et al., 2010) with 25 complete genomes downloaded from the GenBank database (Clark et al., 2016) (Table 2). Nucleoprotein sequences were obtained from the complete genomes with the Bio Edit v7.2 software (Hall et al., 2011). Besides, we added nucleoprotein sequences from the previous study by Streicker et al. (2019) to relate new samples to the previously reported lineages (Streicker et al., 2019). We conducted a phylogenetic analysis using Maximum Likelihood (ML) for the complete genome and nucleoprotein gene, performed the ML trees, and determine the best-fit model in Mega X (Kumar et al., 2018). We accomplished analysis with 1000 bootstrap replicates as statistical support and the substitution models GTR + G + I for complete genomes and T92 + G + I for nucleoprotein gene. Trees generated were also edited with Mega X (Kumar et al., 2018) and used as outgroup the Australian bat virus genome NC_003243.1.
TABLE 2 Genomes used for phylogenetic analysis from Latin American countries
| Host | Country | Access number |
| Australian bat lyssavirus | Australia | NC_003243 |
| Desmodus rotundus | French Guiana | KU523255 |
| Desmodus rotundus | French Guiana | KX148100 |
| Desmodus rotundus | Brazil | KM594041 |
| Desmodus rotundus | Brazil | AB519642 |
| Desmodus rotundus | Brazil | KM594043 |
| Desmodus rotundus | French Guiana | KX148268 |
| Desmodus rotundus | Brazil | KX148109 |
| Desmodus rotundus | Brazil | AB519641 |
| Desmodus rotundus | Brazil | KM594040 |
| Desmodus rotundus | Mexico | JQ685936 |
| Nyctinomops laticaudatus | Brazil | KM594029 |
| Eptesicus furinalis | Brazil | KM594026 |
| Eptesicus furinalis | Brazil | KM594028 |
| Tadarida brasiliensis | Mexico | JQ685963 |
| Dog | Mexico | KX708501 |
| Dog | Mexico | KX708502 |
| Dog | Mexico | KX148102 |
| Dog | Mexico | KX708504 |
| Dog | Mexico | KT006769 |
| Dog | Mexico | KX708503 |
| Tadarida brasiliensis | Argentina | EU293116 |
| Tadarida brasiliensis | Argentina | KX148269 |
| Tadarida brasiliensis | Brazil | KM594038 |
| Tadarida brasiliensis | Brazil | KM594037 |
RESULTS
Optimization of sequencing protocol: Complete viral genome sequencing from clinical samples can be challenging due to the high percentage of host RNA compared to viral RNA and the structural elements intrinsic to viral genomes that hinder sequencing. To improve the coverage of viral genomes and get complete genomes from the study samples, we used a mixture of virus-specific designed primers, random hexamers, and previously published primers (Marston et al., 2013) during the ssDNA generation step; allowing us to obtain complete rabies virus genomes in all cases. However, in this study, the contribution of each set of primers for obtaining complete genomes was not evaluated. The minimum depth of coverage obtained was 68x to sample D03-18. Table 3 shows the assembly statistics.
The length for the isolates D02-18 and D04-18 is 11810 bp, D01-18 is 11809bp, and D03-18 is 11849bp. For all genomes, gene information was complete, the sequences and annotations are available at Genbank (Table 3).
TABLE 3 Assemblage statistics for genomes obtained in Costa Rica
| Samples | Average Depth of coverage | Reads mapped | Genbank accession number |
|---|---|---|---|
| D01-18 | 3 524 x | 274 727 | MW506939 |
| D02-18 | 653 x | 507 68 | MW506938 |
| D03-18 | 68 x | 5 139 | MW506937 |
| D04-18 | 779 x | 70 795 | MW506940 |
Phylogenetic inferences: Genome’s study came from several regions of Costa Rica, as is shown in Figure 1. We performed a preliminary Blast analysis for the scaffolds after assembly, and as expected, the sequences matched only with the rabies virus. Identity analysis showed that three genomes had a similarity of ~99% (D01-18, D02-18, and D04-18) and were isolated in the south and center of the country. In contrast, the isolate D03-18 had a similarity of ~96% with sequences D01-18, D02-18, and D04-18. D03-18 was isolated in the north of the country. These samples are the first reports of complete rabies virus genomes for Costa Rica (Table 3) and do not have significant recombination events found.
We used 25 available genomes in public databases from neighboring countries in America for phylogenetic analyses to study the evolutionary relationships among national isolates and viruses in the Latin American region. Samples D01-18, D02-18, and D04-18 grouped within a monophyletic subclade related to a more phylogenetic distant clade that includes isolate D03-18 and the Mexican sequence JQ685936 (Fig. 2), however the support between these branches is low. Figure 2 shows a division into two large monophyletic groups between rabies transmitted by bats in the wild cycle (in color blue) and rabies transmitted by dogs in the urban cycle reported in Mexico (color red). The second tree (Fig. 3) compared the nucleoprotein sequence against the complete genomes tree used the same type of ML analysis. Both trees showed a similar division and clades, with some differences observed in the bootstrap values for the wild and the urban cycle.
According to Streciker et al. (2019), they found two stationary lineages in Costa Rica toward the north and south of the country, classified as L1 and L2. The L1 lineage is related to outbreaks from South America, and the L2 lineage is related to outbreaks from North America (Streicker et al., 2019). When comparing the national isolates' nucleoproteins against previously reported, we found that samples D01-18, D02-18, and D04-18 are related to the L1 lineage, while the sample D03-18 is related to the L2 lineage (Fig. 3).
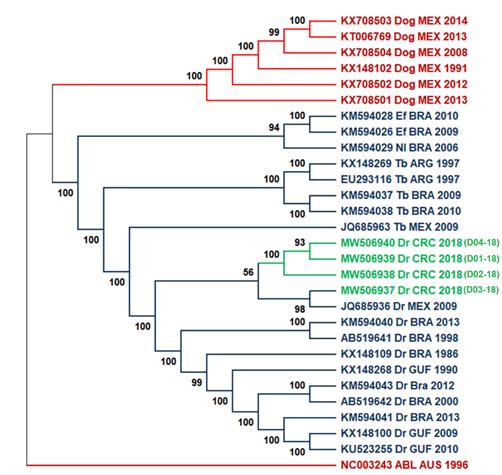
Fig. 2 ML tree of complete genomes using 1000 bootstrap replicates with 25 genome sequences. Costa Rica sequences are highlighted in green. Two principal cluster sequences, in blue, are represented all the virus sequences transmitted by bats (wild cycle), while rabies virus sequences transmitted by dogs (urban cycle) are shown in red. As an outgroup, the Australian bat lyssavirus was used. Lineage abbreviations: Dr - Desmodus rotundus, Tb - Tadarida brasiliensis, EF - Eptesicus furinalis, Nl - Nyctinomops laticaudatus, Dog - canine rabies, ABL - Australian Bat lyssavirus.
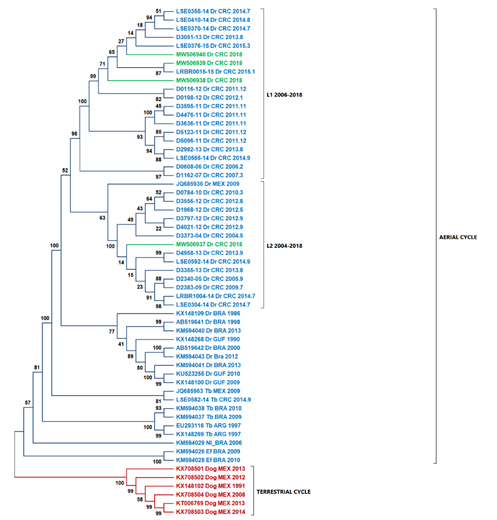
Fig. 3 The ML tree using nucleoprotein sequences and highlighted Costa Rica sequences in green. Two principal cluster sequences, in blue, are represented all the sequences of the virus transmitted by D. rotundus (wild cycle), while in red shown the sequences of rabies virus transmitted by dogs (urban cycle). L1 lineage is formed by sequences coming from South America. L2 lineage is formed by samples from North America. Abbreviations: Dr - Desmodus rotundus, Tb - Tadarida brasiliensis, EF - Eptesicus furinalis, Nl - Nyctinomops laticaudatus, Dog - canine rabies, ABL - Australian
DISCUSSION
Our successful results obtaining complete genomes highlight the applicability of this protocol for obtaining viral genomes from clinical specimens with no need for extensive virus manipulation for enrichment, such as cell culture or ultracentrifugation. However, we did not evaluate in detail the effect of the virus-specific primers compared with random primers in the generations of copy DNA as other factors influence the preparation of genomic libraries. As observed before (Houldcroft et al., 2017), adequate enrichment strategies are preferred to obtain whole viral genomes from primary samples and optimize recovery percentages.
The average depth of coverage obtained using our strategy allowed obtaining consensus sequences for further phylogenetic analysis and is comparable to other reported methodology for sequencing small genomes using Illumina technology (Desai et al., 2013). The lowest coverage value obtained is 68X and can be explained by an intrinsic minimal viral load in the sample or some degradation process. However, samples were adequately preserved and manipulated in nuclease-free conditions; also, we conducted quality controls on total RNA.
A large amount of host mRNA is obtained in proportion to the viral interest RNA (Oyola et al., 2013), and there is a decrease in both RNAs when the depletion is performed (Marston et al., 2013). Therefore, virus-specific primers improve genome coverage (Marston et al., 2013), and we implemented them in our protocol (Table 3).
When analyzing the grouping of all samples, two monophyletic clades are seen (Fig. 2). Isolates from the wild cycle formed the first clade, which contains the study samples. The dog-borne virus forms the second in the canine rabies cycle. The clustering observed in figures 2 and 3 for all clades corresponds with the previously reported in the literature (Delmas et al., 2008; Garcés-Ayala et al., 2017; Kuzmin et al., 2012; Lavergne et al., 2016; Mochizuki et al., 2011). The tree topology obtained in the ML tree coincided with that obtained for the nucleoprotein gene tree in figure 3, which was expected considering the best resolution obtained with complete sequences against the nucleoprotein gene, even though some branches not having adequate support at nodes (>70%) (Soltis & Soltis, 2003). Similar topologies were observed in these trees and can be due to the lower number of sequences analyzed. However, we expected NGS analysis to provide better resolution and inferences than nucleoprotein sequences since NGS can detect a broader spectrum of mutations (Bankevich et al., 2012; Capobianchi et al., 2013).
The phylogenetic characterization of rabies genomes and the nucleoprotein sequences demonstrated all new isolates related to the rabies virus transmitted by D. rotundus, as in previous studies in Costa Rica (Badilla et al., 2003; Hutter et al., 2016; Streicker et al., 2019). These data evidenced that the two-lineage virus described previously (Streicker et al., 2019) is still circulating in Costa Rica. The vampire D. rotundus (Almeida et al., 2005; Calderón et al., 2019; Ellison et al., 2014; Escobar et al., 2015) is the most significant vector that mainly infects cattle (Brito-Hoyos et al., 2013; Cargnelutti et al., 2017) but no evidence of the terrestrial cycle is observed. Typically, annual outbreaks of rabies are observed in the depicted regions of the country. However, another significant outbreak hotspot is the North Atlantic region of the country. During 2018, there were 20 rabies outbreaks distributed in the provinces of Alajuela, San Jose, and Puntarenas but, no bovine paralytic rabies cases were reported in the North Atlantic region, which coincides with previously reported by Streicker et al. (2019) for the region (Streicker et al., 2019).
Genomes phylogenetic analysis and nucleoprotein sequence show that isolate D03-18 did not cluster in the same subclade as isolates D01-18, D02-18, and D04-18. The differences observed in isolate D03-18 with the other national isolates can be due to adaptations in the evolutionary dynamics of the virus lineages in D. rotundus (Brunker et al., 2018; de Almeida Campos et al., 2020) based on the spread of the virus strain in bat colonies (León et al., 2021; Streicker et al., 2019).
Phylogenetic analysis based exclusively on the nucleocapsid region allows us to contextualize the isolates of this study in comparison with the lineages L1 and L2 determined previously (Streicker et al., 2019). Streicker et al. (2019) described two lineages in Costa Rica. Samples D01-18, D02-18, and D04-18 grouped in the same clade belonging to L1. Sample D03-18 was grouped closer to JQ685936 (Mexico) within L2, which explains the differences between D03-18 and the other isolates from the southern and central parts of the country. This result is consistent with the differences in the percentage of identity obtained. Also, our analysis did not suggest recombination as a source of variability within the new genomes described in this study.
In conclusion, our study describes the first-time complete genomes of rabies virus reported in Costa Rica and contextualized the latest isolates with the Lineage classification previously proposed by Streicker et al. (2019). This result provides some insights into potential transmission sources based on phylogeography. Future comparisons will focus on tracing the evolutionary changes of the rabies virus in the region in greater detail.

