











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La cuantificación con emisión atómica, se da cuando los átomos se relajan a sus niveles de energía basales después de ser excitados, cediendo su exceso de energía en forma de fotones de luz o bien intensidad. Esta técnica tiene diversas aplicaciones en el análisis del sodio (Na), por ejemplo, se analiza en los campos analíticos de la biología, la agricultura, entre otros, además es de gran interés en el análisis de fluidos biológicos y en tejidos [1]. En esta técnica se puede medir la concentración de un elemento mediante la intensidad de la llama, ya que existe una correlación entre estas dos variables. Además, se generan resultados cuantitativos con alta reproducibilidad, debido a que los espectros obtenidos en los análisis realizados son espectros sencillos, los cuales no presentan interferencias espectrales [2 ].
Las técnicas instrumentales presentan menos fuentes de error que las técnicas de química húmeda. Por tanto, se disminuye en un alto grado el error sistemático del analista [3], [4]. Además, la exactitud y la precisión del mismo mejoran con respecto a la química húmeda [5]. Con este método, se garantiza que las soluciones de estudio están siendo monitoreadas de manera correcta, lo cual es importante debido a que la inyección de cloruro de sodio (NaCl) al 0,9% y la inyección de dextrosa al 5% con NaCl al 0,9%, son productos solicitados en las diversas clínicas y hospitales de Costa Rica; ya que estas cumplen con el control de la distribución del agua en el organismo y mantiene el equilibrio de líquidos [6].
El NaCl es una materia prima importante en la industria química, puesto que se le da diversos usos al mismo [7]. En el laboratorio implicado en el estudio, se utiliza el NaCl como materia prima, para fabricar soluciones parenterales, tales como: inyección de NaCl al 0,9% e inyección de dextrosa al 5% con NaCl al 0,9% (solución mixta). Las cuales, cumplen gran importancia en el campo de la salud, son utilizadas en terapias de rehidratación en los casos de diarrea aguda y cólera., también para el traumatismo, las quemaduras, cuando los pacientes poseen un déficit de Na corporal y controlan la distribución del agua en el organismo [6].
En el contexto analítico, según la Farmacopea de Estados Unidos de América (USP) [8] la determinaciónde NaCl en las soluciones parenterales NaCl al 0,9% y la solución mixta, se realiza mediante una titulación volumétrica, la cual utiliza nitrato de plata (AgNO3) y Eosina “Y” como indicador. La USP establece el uso de la técnica de emisión para soluciones parenterales complejas. Según la investigación bibliográfica realizada no hay estudios en donde se haya utilizado la emisión atómica para la determinación de Na en soluciones parenterales.
No obstante, se analiza el Na mediante emisión atómica en otro tipo de muestras, por ejemplo, Osicka y Giménez [9], realizaron un estudio de determinación de Na y potasio (K) en aguas naturales subterráneas, donde se utilizó la metodología descrita de la Asociación Oficial de Química Analítica (AOAC sus siglas en inglés). Por otra parte, Montero et al, [10] determinaron la cantidad de Na en panes y snacks de mayor consumo en Costa Rica, según la AOAC. Y en el estudio de Alvarado y Peñaloza [11], se realizó la determinación de la sensibilidad y especificidad en métodos diferentes para el análisis de Na y K en suero humano, dentro de estos, se analizó la emisión atómica.
En el laboratorio involucrado se determina el Na de manera indirecta, el método empleado es una modificación del método de la Farmacopea, en el cual se utiliza AgNO3 como titulante y diclorofluoresceína como indicador, el método a desarrollar busca eliminar el uso del AgNO3, porque este puede producir lesiones pulmonares y renales, además puede causar argiria, la cual se trata de una coloración oscura en la piel debido al contacto del reactivo con la piel, también puede causar reacciones alérgicas [12].
Fundamento
La espectroscopia de emisión atómica, analiza las longitudes de onda de los fotones que se emiten por los átomos durante la transición desde el estado excitado a un estado de menor energía. Para lograr que los átomos se encuentren en un estado excitado, se calienta la muestra a temperatura alta a través de una llama, esto produce excitaciones como consecuencia de las colisiones entre los átomos de la muestra cuando adquieren energía debido a la llama [13], [14]. Esta técnica analítica, es diferente a la técnica de espectroscopia de absorción atómica, debido a que en la emisión atómica la fuente de generación de la emisión de la radiación es la propia materia a analizar [15].
La fotometría de llama se utiliza casi exclusivamente para la determinación de Na y K. Esto debido a que los metales alcalinos son elementos que llegan al estado de excitación con facilidad en la llama, además, emiten energía a longitudes de onda específicas y diferentes a los demás [16], [17]. En este método se determina indirectamente la cantidad de NaCl, compuesto vital para la nutrición y para los procesos fisiológicos del cuerpo humano [18]. Las soluciones parenterales son importantes en el campo de la salud, ya que son utilizadas en terapias de rehidratación y en reposición de los electrolitos [19]. La solución de dextrosa al 5% y NaCl al 0,9% es utilizada para proveer electrolitos a los pacientes con un déficit de los mismos.
Es importante el monitoreo de la cantidad de NaCl presente en las soluciones parenterales ya que, si se tiene una sobredosis de este, se puede producir acidosis hiperclorémica, que causa acidemia a nivel sistémica, tisular y celular en el cuerpo [20]. La administración de la solución parenteral es importante en caso de que existan déficits de Na en el cuerpo y el cuerpo necesita que haya un equilibrio entre el Na y el K [21].
Metodología
Pruebas con controles
Se tomaron tres controles correspondientes al patrón de menor concentración, el de concentración media y el de mayor concentración de la curva de calibración estándar, esta se preparó a partir de un patrón estándar de Na (10074 ppm), con seis niveles de concentración correspondientes a 0,80 - 1,0 - 1,20 - 1,40 - 1,60 y 1,80 g/L. Para la medición se toman alícuotas de 100 µL a partir de los patrones preparados y una alícuota de 10,00 mL de diluyente concentrado y se mide la intensidad en el fotómetro. Se midió la curva de calibración estándar, y se midió cuatro veces cada control. Y se realizó una comparación de medias experimentales.
Comparación entre el método fotométrico y el método oficial de la USP 38
Se ejecutó un análisis de 10 muestras de un mismo lote (para ambos productos) por medio de los dos métodos y al obtener los resultados correspondientes se hizo una comparación de medias experimentales. En el método para determinación de NaCl mediante el método oficial de la USP 38, se siguió lo especificado en este. En el método fotométrico para la determinación de Na: se preparó una curva de calibración (preparación igual que para pruebas con controles). Para las muestras se tomó una alícuota de 8,30 mL de la solución en un balón de 25,00 mL y se aforó con agua destilada.
Validación del método
Con el apoyo de la guía para validación de métodos del ECA [22],se utilizaron dos soluciones parenterales (inyección de NaCl al 0,9% y la inyección de dextrosa al 5% y NaCl al 0,9%), se trabajó un mismo lote para cada solución para las pruebas de validación.
Linealidad
Se prepararon tres curvas de calibración independientes a partir de un estándar de Na (10074 ppm), la preparación es igual que para pruebas con controles. Se realizó la linealidad del método, donde se prepararon tres curvas de calibración independientes de los dos productos mencionados, en este caso se trabajaron concentraciones correspondientes a 0,87 - 1,08 - 1,30 - 1,51 y 1,73 g/L.
Repetibilidad y precisión intermedia
Para la repetibilidad del sistema se preparó un patrón con concentración de 1,40 g/L y se midió un total de seis veces, con respecto a la repetibilidad del método se realizaron seis preparaciones independientes del patrón de concentración de 1,40 g/L y se midieron. Para la precisión intermedia se preparó un patrón de 1,40 g/L y este se midió seis veces durante seis distintos días.
En relación a la repetibilidad y precisión intermedia de cada producto, se prepararon seis patrones de 1,30 g/L, para ambos productos y para la precisión intermedia se preparó un patrón de cada producto cada día por seis días.
Exactitud
Para la evaluación de la exactitud se realizó una comparación de los porcentajes de recuperación con respecto al valor verdadero de los productos estudiados.
Resultados
En el Cuadro 1 se pueden observar los desvíos estándar relativos (DSR%) obtenidos para las mediciones de los controles, los cuales son ≤ 2%.
Cuadro 1 Resultados obtenidos para las pruebas de desempeño del fotómetro.
| - | Medición 1 (g/L) | Medición 2 (g/L) | Medición 3 (g/L) | Medición 4 (g/L) | Promedio | DSR % |
|---|---|---|---|---|---|---|
| Control 1 | 0,81 | 0,79 | 0,81 | 0,79 | 0,80 | 1,51 |
| Control 2 | 1,41 | 1,37 | 1,41 | 1,39 | 1,40 | 1,46 |
| Control 3 | 1,78 | 1,74 | 1,77 | 1,77 | 1,76 | 1,11 |
Seguidamente, el en Cuadro 2 y en el Cuadro 3 se evidencia los resultados de las pruebas comparativas del método fotométrico implementado y el método oficial de la USP 38, para el producto 1 (NaCl al 0,9%) y el producto 2 (solución mixta) respectivamente.
En lo referente a la validación del método, se muestra en la Figura 1, 2 y 3, la superposición de las tres curvas de calibración correspondientes a las curvas preparadas para la validación del parámetro de linealidad del método, del producto 1 y del producto 3, respectivamente, en estas se obtuvieron que los coeficientes de correlación (R2) son valores mayores a 0,995.
En el Cuadro 4 se muestra la concentración obtenida para la repetibilidad del sistema, además se muestra la desviación estándar (DS) y el DSR% de las mediciones obtenidas y en el Cuadro 5 se muestra la repetibilidad de ambos productos.
Cuadro 2 Análisis ANOVA del método USP y método fotométrico para el Producto 1.
| Prueba t para dos muestras suponiendo varianzas iguales | ||
|---|---|---|
| - | Varible 1 | Variable 2 |
| Media | 97.29177778 | 97.02823946 |
| Varianza | 0.035136148 | 1.09880885 |
| Observaciones | 3 | 5 |
| Varianza agrupada | 0.744251282 | - |
| Diferencia hipotética de las | 0 | - |
| Grados de libertad | 6 | - |
| Estadístico t | 0.418296867 | - |
| P(T<=t) una cola | 0.345144387 | - |
| Valor crítico de t (una cola) | 1.943180281 | - |
| P(T<=t) dos colas | 0.690288775 | - |
| Valor crítico de t (dos colas) | 2.446911851 | - |
Cuadro 3 Análisis ANOVA del método USP y método fotométrico para el Producto 2.
| Prueba t para dos muestras suponiendo varianzas iguales | ||
|---|---|---|
| - | Varible 1 | Variable 2 |
| Media | 97.61644444 | 97.9856411 |
| Varianza | 0.140544593 | 0.508290738 |
| Observaciones | 3 | 5 |
| Varianza agrupada | 0.38570869 | - |
| Diferencia hipotética de las | 0 | - |
| Grados de libertad | 6 | - |
| Estadístico t | -0.814008023 | - |
| P(T<=t) una cola | 0.223367187 | - |
| Valor crítico de t (una cola) | 1.943180281 | - |
| P(T<=t) dos colas | 0.446734374 | - |
| Valor crítico de t (dos colas) | 2.446911851 | - |
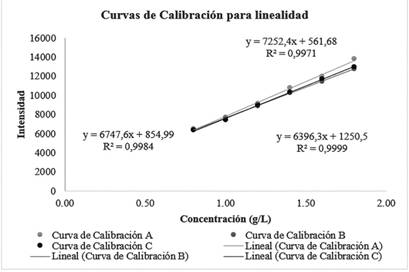
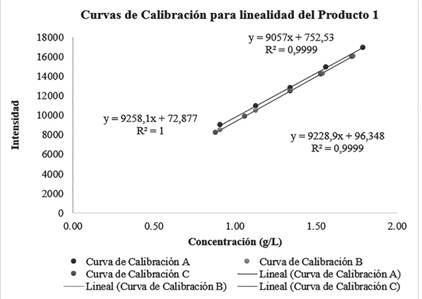
Figura 2 Superposición de curvas de calibración para comprobar linealidad del método en relación al producto inyección de cloruro de sodio al 0,9%.
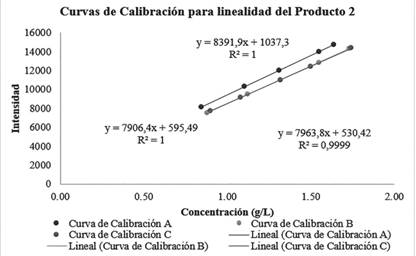
Figura 3 Superposición de curvas de calibración para comprobar linealidad del método en relación al producto inyección de dextrosa al 5% y cloruro de sodio al 0,9%.
Cuadro 4 Resultados de la validación del parámetro de repetibilidad del sistema.
| Repetición | Concentración teórica (g/L) | Concentración experimental (g/L) | DS | DSR% | Aceptación |
|---|---|---|---|---|---|
| Repetibilidad del Sistema | |||||
| 1 | - | 1,40 | - | - | - |
| 2 | - | 1,40 | - | - | - |
| 3 | 1,40 | 1,38 | 0,01 | 0,69 | ≤2% |
| 4 | - | 1,38 | - | - | - |
| 5 | - | 1,38 | - | - | - |
| 6 | - | 1,38 | - | - | - |
Cuadro 5 Resultados de la validación del parámetro de repetibilidad del método.
| Repetición | Concentración teórica (g/L) | Concentración experimental (g/L) | DS | DSR% | Aceptación |
|---|---|---|---|---|---|
| Repetibilidad del Producto 1 | |||||
| 1 | - | 1,34 | - | - | - |
| 2 | - | 1,32 | - | - | - |
| 3 | 1,30 | 1,32 | 0,01 | 1,01 | ≤2% |
| 4 | - | 1,32 | - | - | - |
| 5 | - | 1,31 | - | - | - |
| 6 | - | 1,30 | - | - | - |
| Repetibilidad del Producto 2 | |||||
| 1 | - | 1,34 | - | - | - |
| 2 | - | 1,32 | - | - | - |
| 3 | 1,30 | 1,32 | 0,02 | 1,52 | ≤2% |
| 4 | - | 1,32 | - | - | - |
| 5 | - | 1,31 | - | - | - |
| 6 | - | 1,30 | - | - | - |
En el Cuadro 6 se muestran los resultados de la precisión intermedia para el sistema y en el Cuadro 7 se muestran los resultados para precisión intermedia del método, ensayada con ambos productos (1 y 2).
Cuadro 6 Resultados de la validación del parámetro de precisión intermedia del sistema.
| Repetición | Concentración teórica (g/L) | Concentración experimental (g/L) | DS | DSR% | Aceptación |
|---|---|---|---|---|---|
| 1 | - | 1,41 | - | - | |
| 2 | - | 1,39 | - | - | - |
| 3 | 1,40 | 1,41 | 0,01 | 0,70 | ≤2% |
| 4 | - | 1,41 | - | - | - |
| 5 | - | 1,40 | - | - | - |
| 6 | - | 1,39 | - | - | - |
Cuadro 7 Resultados de la validación del parámetro de precisión intermedia del método.
| Repetición | Concentración teórica (g/L) | Concentración experimental (g/L) | DS | DSR% | Aceptación |
|---|---|---|---|---|---|
| Precisión intermedia del Producto 1 | |||||
| 1 | - | 1,34 | - | - | - |
| 2 | - | 1,36 | - | - | - |
| 3 | 1,30 | 1,32 | 0,02 | 1,37 | ≤2% |
| 4 | - | 1,34 | - | - | - |
| 5 | - | 1,32 | - | - | - |
| 6 | - | 1,33 | - | - | - |
| Precisión intermedia del Producto 2 | |||||
| 1 | - | 1,34 | - | - | - |
| 2 | - | 1,38 | - | - | - |
| 3 | 1,30 | 1,32 | 0,02 | 1,59 | ≤2% |
| 4 | - | 1,35 | - | - | - |
| 5 | - | 1,33 | - | - | - |
| 6 | - | 1,33 | - | - | - |
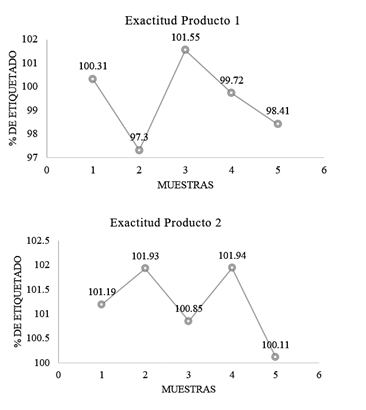
En el Cuadro 8, se incluye los resultados obtenidos para la exactitud del método, se tomó como referencia el valor verdadero de 101,32% y 100,24% para el producto 1 y para el producto 2 respectivamente.
Por parte de la especificidad, los blancos preparados obtuvieron intensidades menores que la intensidad del primer patrón (Cuadro 9).
Por otra parte, se realizó el análisis a tres diferentes lotes de cada producto en estudio y de estos, cada lote se analizó por triplicado; los resultados obtenidos se aprecian en el Cuadro 10 para el producto 1 y en el Cuadro 11 para el producto 2.
Cuadro 8 Resultados de la validación del parámetro de exactitud de los productos de estudio.
| Datos | % Etiquetado Obtenido | Valor de Referencia | % de Recuperación | Parámetro de Aceptación |
|---|---|---|---|---|
| Exactitud Producto 1 | ||||
| 1 | 101,63 | - | 100,31 | - |
| 2 | 98,59 | - | 97,30 | - |
| 3 | 102,89 | 101,32 | 101,55 | 97-103 % |
| 4 | 101,03 | - | 99,72 | - |
| 5 | 99,71 | - | 98,41 | - |
| Promedio | 100,77 | - | - | - |
| DSR % | 1,66 | - | - | - |
| Exactitud Producto 2 | ||||
| 1 | 101,44 | - | 101,19 | - |
| 2 | 102,18 | - | 101,93 | - |
| 3 | 101,10 | 100,24 | 100,85 | 97-103 % |
| 4 | 102,18 | - | 101,94 | - |
| 5 | 100,35 | - | 100,11 | - |
| Promedio | 101,45 | - | - | - |
| DSR % | 0,76 | - | - | - |
Cuadro 9 Resultados de la validación del parámetro de especificidad.
| Blanco | Intensidad obtenida | Intensidad Primer Patrón |
|---|---|---|
| Dextrosa al 5% | 581 | - |
| Agua | 578 | 6517 |
| Agua y Dextrosa al 5% | 574 | - |
Cuadro 10 Resultados de la concentración obtenida en tres lotes diferentes del producto 1.
| Muestra | Concentración (g/L) | %Etiquetado | Promedio de Etiquetado | DSR % |
|---|---|---|---|---|
| Lote L39R5 | ||||
| 1 | 9,10 | 101,08 | - | - |
| 2 | 9,14 | 101,51 | 101,05 | 0,46 |
| 3 | 9,05 | 100,57 | - | - |
| Lote L39S2 | ||||
| 1 | 9,08 | 100,85 | - | - |
| 2 | 9,06 | 100,65 | 100,49 | 0,47 |
| 3 | 9,00 | 99,95 | - | - |
| Lote L39R9 | ||||
| 1 | 9,11 | 101,21 | - | - |
| 2 | 9,06 | 100,71 | 100,90 | 0,27 |
| 3 | 9,07 | 100,77 | - | - |
Cuadro 11 Resultados de la concentración obtenida en tres lotes diferentes del producto 2.
| Muestra | Concentración (g/L) | %Etiquetado | Promedio de Etiquetado | DSR % |
|---|---|---|---|---|
| Lote L39R5 | ||||
| 1 | 9,06 | 100,64 | - | - |
| 2 | 8,99 | 99,86 | 100,20 | 0,40 |
| 3 | 9,01 | 100,12 | - | - |
| Lote L39R0 | ||||
| 1 | 8,98 | 99,78 | - | - |
| 2 | 8,96 | 99,61 | 99,80 | 0,21 |
| 3 | 9,00 | 100,02 | - | - |
| Lote L37X1 | ||||
| 1 | 9,35 | 103,88 | - | - |
| 2 | 9,30 | 103,35 | 103,59 | 0,26 |
| 3 | 9,32 | 103,55 | - | - |
Discusión
Como se puede observar en el cuadro 1, los resultados obtenidos para los controles aportan que no existe variabilidad en las mediciones realizadas, esto se demuestra por el DSR% obtenido para cada uno de los controles, los cuales son ≤ 2%, criterio de aceptación establecido en este caso. Por otra parte, en la comparación entre el método fotométrico y el método oficial de la USP 38, se obtuvo que los datos que arroja el método de emisión son estadísticamente comparables con los resultados obtenidos por el método oficial (ver cuadros 2 y 3), debido a que el estadístico t obtenido en ambos casos son menores que el valor crítico de t. Lo cual demuestra que el método fotométrico aporta resultados confiables que son estadísticamente iguales a los que brinda el método oficial.
En cuanto a lo obtenido en la validación del método se muestra en la figura 1 los datos correspondientes a la linealidad del sistema, en donde se observan las curvas de calibración obtenidas por triplicado para el sistema a diferentes niveles de concentración, se observa que las tres curvas arrojan ecuaciones de la recta distintas, no obstante, mantienen valores cercanos (excepto por la Curva B). Además, las tres curvas cuentan con una regresión deseable y existe una correlación significativa, puesto que los R2 obtenidos para las tres curvas son mayores a 0.995 el cual es el criterio de aceptación que dicta el Ministerio de Salud.
Para la evaluación de este parámetro, se evaluó también la homocedasticidad, puesto que para que un método sea lineal el mismo debe presentar un comportamiento homocedástico [23], [24]. La estimación de la homocedasticidad se realizó mediante el uso del test de Cochran, el cual se calculó mediante la siguiente ecuación:
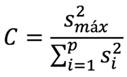
Al calcular este valor C, el mismo se compara con los valores críticos establecidos para la prueba de Cochran, para el caso de la homocedasticidad del sistema (Figura 4), se obtuvo un valor C de 0,59 el cual es menor que el valor crítico 0,8772, lo cual demuestra que las varianzas son iguales en el caso de las tres curvas preparadas, es decir que existe un comportamiento homocedástico.
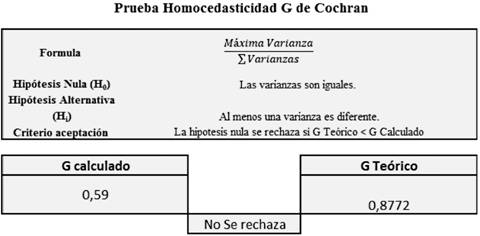
Seguidamente, se muestra un análisis de los residuos (Figura 5) correspondientes a la validación de la linealidad del sistema, en este no se presentan tendencias sistemáticas, es decir, se presenta un comportamiento aleatorio, lo cual indica la linealidad [23].
Seguidamente, se muestra un análisis de los residuos (Figura 5) correspondientes a la validación de la linealidad del sistema, en este no se presentan tendencias sistemáticas, es decir, se presenta un comportamiento aleatorio, lo cual indica la linealidad [23].
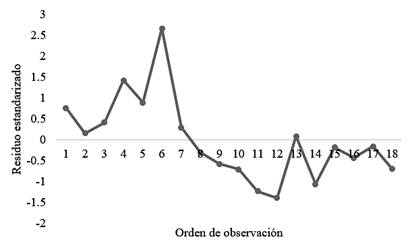
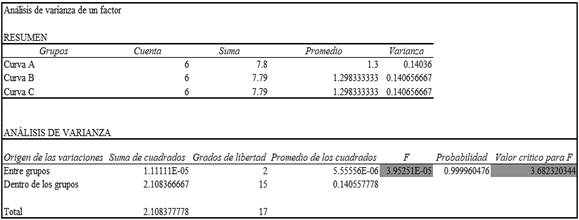
De acuerdo al análisis de ANOVA realizado en Excel (Figura 6), se obtuvo un valor de F de 3,9x10-5 el cual es ≤ que el F crítico 3,68. Por tanto se demuestra que las varianzas de las tres curvas de calibraciónno varían entre ellas.
Con respecto a linealidad relacionada al producto 1 (NaCl 0.9%), se incluye en la figura 2 los datos obtenidos, en esta se observan las curvas de calibración obtenidas por triplicado, las tres curvas arrojan ecuaciones de la recta distintas, sin embargo, los valores de las ecuaciones se mantienen similares. Se cuenta con una regresión deseable y existe una correlación significativa, debido a que los R2 obtenidos para las tres curvas cumplen con el criterio de aceptación que establece el Ministerio de Salud (0.995).
Se realizó también la evaluación de la homocedasticidad, de la misma manera en que se mencionó anteriormente, en este caso se obtuvo un valor C de 0,31 el cual es menor que el valor crítico 0,9057, lo cual demuestra que existe un comportamiento homocedástico. En el análisis de los residuos correspondientes a la validación de la linealidad del producto 1, los residuos mantienen un comportamiento aleatorio alrededor de la línea central, por tanto, existe linealidad. Conforme al análisis de ANOVA realizado en Excel para los datos de linealidad del producto 1, se obtuvo un valor de F calculado de 0,017el cual es menor que el valor de F crítico 3,88. Este resultado evidencia que las varianzas de las tres curvas de calibración del producto 1 no difieren entre ellas.
En relación a la linealidad del producto 2 (NaCl al 0.9% y Dextrosa a 5%), las tres curvas (Figura 3) arrojan ecuaciones de la recta distintas, sin embargo, los valores de las ecuaciones se mantienen similares. Las curvas de calibración cuentan con una regresión deseable y existe una correlación significativa, puesto que los R2 son mayores a 0.995. Se evaluó la homocedasticidad, de la misma manera que se realizó para los casos anteriores, se obtuvo un valor C de 0,31 el cual es menor que el valor crítico 0,9057, lo cual demuestra que existe un comportamiento homocedástico.
Consecutivamente, en el análisis de los residuos correspondientes a la validación de la linealidad del producto 2, se asegura que los residuos mantienen un comportamiento aleatorio, es decir que se demuestra que existe linealidad. Finalmente, en el análisis de ANOVA realizado en Excel para los datos de linealidad del producto 2, se obtuvo un valor de F calculado de 0,011 el cual es menor que el valor de F crítico 3,88. Lo cual evidencia que las varianzas de las tres curvas de calibración del producto 2 no difieren entre ellas.
En relación a los resultados de repetibilidad (cuadros 4 y 5), se puede observar que ninguno de los DSR% obtenidos para el sistema y el método en ambos productos sobrepasa el parámetro establecido por el Ministerio de Salud para el contenido de principio activo el cual corresponde a un 2% de DSR. Por tanto, estos resultados evidencian que el método cuenta con una precisión óptima. Con respecto a la precisión intermedia (cuadros 6 y 7), la variable que se cambió fue el día, por tanto, se mantuvieron las variables: analista y equipo constantes y se observa que el DSR % fue ≤ 2% por tanto, se demuestra que, aunque el análisis se realiza en días diferentes, el mismo otorga resultados que no varían abruptamente.
Por parte de la exactitud (cuadro 8) ninguno de los datos del porcentaje de recuperación sobrepasa el parámetro máximo de aceptación de 97-103 %, lo cual se evidencia en la figura 7. Esto demuestra que el método brinda resultados exactos y confiables.
En el caso del parámetro de especificidad (Cuadro 9), las señales obtenidas para los blancos preparados no interfieren con el análisis, puesto que las señales obtenidas son mucho menores a la señal obtenida para el primer patrón, esto demuestra que los otros componentes de la muestra analizada no interfieren en la medición del analito de interés.
En lo que respecta al monitoreo con muestras, en el Cuadro 10 se presentan los resultados obtenidos para el producto 1 (NaCl 0.9%), en el mismo se observa que los tres lotes analizados presentan un DSR % idóneo, debido a que fue ≤ 2%, además el porcentaje de lo etiquetado para los tres lotes es aceptable, debido a que el parámetro de aceptación es de 90-110% por tanto las muestras analizadas cumplen con lo establecido por la farmacopea.
En el Cuadro 11, se incluyen los resultados obtenidos para el producto 2 (NaCl 0.9% y Dextrosa 5%), en este se observa que los tres lotes analizados presentan DSR% cercanos a 0, lo cual es un buen indicio de la precisión del análisis. Además, el porcentaje de etiquetado no sobre pasa el parámetro que establece la USP (90-110%). Esta solución se recomienda a los pacientes que no pueden utilizar la vía oral para administrar un aporte calórico y los requerimientos de Na, glucosa y agua al cuerpo. Por tanto, es deseable que se encuentre con un etiquetado aceptable [25].
Conclusiones
Mediante la prueba estadística de T, se obtuvo que el método fotométrico aporta resultados que son estadísticamente semejantes a los resultados brindados por el método oficial USP. Por lo tanto, se tiene que los métodos son comparables estadísticamente.
La validación de un método no oficial es necesaria, ya que de esta manera se garantiza la eficiencia y eficacia de la implementación del método. En la validación desarrollada en este estudio se obtuvo que las pruebas realizadas cumplieron con los criterios de aceptación, por tanto, se obtuvo evidencia estadística que indica que el método validado aporta resultados confiables, exactos y precisos para la determinación de Na en soluciones parenterales.
De acuerdo con el análisis realizado a los distintos lotes de cada producto, se tiene que los productos elaborados por el laboratorio implicado, cumplen con las especificaciones, puesto que se encuentran dentro del criterio de aceptación establecido por la Farmacopea.

