











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
PermalinkIntroducción
Una de las zonas con mayor riqueza de especies en el mundo es el Chocó biogeográfico, donde se encuentran ampliamente distribuidos los bosques húmedos tropicales que albergan una gran diversidad de mirmecofauna (Fowler et al., 1989). En esta región del Pacífico Oriental Tropical (POT sensu latto) (Robertson & Allen, 2015; Vega, Hernandez-Guerrero, & Cruz-Barraza, 2012) el conocimiento sobre la distribución de la hormiga cortadora Atta cephalotes, así como la variación genética de la especie en el Pacífico colombiano y su relación con agrupaciones poblacionales propuestas en estudios previos es limitado. Se ha propuesto que A. cephalotes conforma dos grupos genéticamente estructurados en el Neotrópico, un grupo se encuentra distribuido en Centroamérica y el noroccidente de los Andes colombianos, y un segundo grupo se extiende desde los Andes ecuatorianos hasta el sur de la Amazonía en Brasil (Solomon, Bacci, Martins, Goncalves Vinha, & Mueller, 2008); una hipótesis biogeográfica que aún debe ser examinada.
La hormiga cortadora Atta cephalotes es uno de los organismos herbívoros más exitoso del Neotrópico con una amplia distribución desde México hasta Argentina (Bacci et al., 2009; Solomon, 2007). Estudios previos sugieren que las hormigas cortadoras habitan islas del Caribe, las Antillas, así como islas del Pacífico Oriental (Haines, 1975; Solomon, 2007; Terborgh, Feeley, Silman, Nuñez, & Balukjian, 2006; Valdés-Rodríguez, Chacón De Ulloa, Armbrecht, 2014). Sin embargo, la ocurrencia y dispersión de A. cephalotes en áreas insulares del Pacífico ha sido poco estudiada a pesar de ser un componente común de la mirmecofauna (Lincango & Morales, 2005; Solomon & Mikheyev, 2005; Solomon, 2007; Valdés-Rodríguez et al., 2014). Asimismo, aspectos del proceso de colonización y de variación genética de estas poblaciones insulares son prácticamente desconocidos.
La Isla Gorgona, una de las dos áreas insulares del Pacífico colombiano donde predomina la selva húmeda tropical (UAESPNN, 2018), presenta una alta riqueza de hormigas y A. cephalotes se destaca como una de las hormigas más comunes en la isla (Valdés-Rodríguez et al., 2014). Aunque existen diferentes estudios sobre ecología, diversidad y riqueza de hormigas (Arcusa, 2017; Vázquez-Moreno, Matienzo, Alfonso, Moreno, & Álvarez, 2009; Zabala, Arango, & Chacón de Ulloa, 2013), el conocimiento sobre la sistemática de las hormigas cortadoras, así como aspectos biológicos y ecológicos de varias especies es aún insuficiente (Fernández, 2003). Actualmente se desconoce el patrón de distribución espacial de la variación genética de A. cephalotes para Colombia, incluyendo la Isla Gorgona.
Este estudio tiene como objetivo realizar la caracterización del gen Citocromo Oxidasa I de A. cephalotes para examinar su variación en la Isla Gorgona en el marco del POT. Con este marcador se examinó si la diversidad genética de A. cephalotes en la Isla Gorgona está cercanamente relacionada con la región oriental de la cordillera de los Andes (región cis-Andina), como supone la propuesta biogeográfica de Solomon et al. (2008). Para esto se implementó un análisis de varianza molecular, así como un análisis bayesiano de asignación de individuos en poblaciones, y utilizando un método basado en la teoría de circuitos se calculó un mapa de conectividad genética de A. cephalotes en el POT. Además, mediante un análisis de la coalescencia, se examina si la separación entre la Isla Gorgona y el margen continental del Pacífico oriental representa una barrera que podría haber facilitado la estructuración genética de A. cephalotes.
Materiales y métodos
Área de estudio y recolección de muestras: Este estudio se realizó en el Parque Nacional Natural Isla Gorgona ubicado en el Pacífico colombiano (2°9’ N & 78°2’ W) a 35 km de distancia desde el punto más cercano a la costa. El parque cuenta con un área terrestre de 1 382.29 Ha y su altura máxima se encuentra a 338 m.s.n.m. (UAESPNN, 2018). La recolección de individuos en este sitio se realizó durante caminatas a lo largo del borde oriental de la isla como en el borde sur-occidental; al interior del bosque, entre la zona de asentamiento (al sur-occidente de la isla) y sobre el camino hacia el punto más alto de la isla. Adicionalmente, se incluyen muestras secuenciadas de novo para localidades del Pacífico colombiano como Quibdó, Buenaventura, Dagua y Guapi.
PCR y secuenciación de ADN: El marcador genético utilizado para este estudio fue la subunidad I del gen mitocondrial que codifica para la enzima Citocromo Oxidasa (COI). La extracción de ADN genómico se realizó con el kit Thermo Scientific®, para la amplificación del gen COI por una reacción en cadena de la polimerasa (PCR). Se utilizó 3.5 µl de H2O, 12.5 µl de Taq DNA polimerasa (1X One Taq Hot Start Quic Maxter Mix), 2.5 µl BSA, 0.5 µl DMSO, 0.5 µl de cada cebador, 5 ng de DNA para un volumen total de 25 µl. Los cebadores utilizados fueron GCTACTACATAATAKGTATCATG (Kronauer, Gadau, & Hölldobler, 2004) y CAACATTTATTTTGATTTTTTGG (Simon et al., 1994). Para el PCR se realizó una denaturación inicial a 94 ºC (4 min) posteriormente una más corta a 94 ºC durante 40 s, un alineamiento a 45 ºC (1 min), elongación a 72 ºC (1.5 min) por 6 ciclos desde el segundo paso. Luego una segunda denaturación a 94 ºC (1 min), una alineación a 55 ºC (1 min), una elongación a 72 ºC (1.5 min) por 30 ciclos y una extensión final a 72 ºC (10 min). Los productos de la PCR fueron separados por electroforesis en un gel de agarosa de 2 % y posteriormente se enviaron a Macrogen Inc. para su purificación y secuenciación usando una reacción con BigDye (Applied BiosystemsTM).
La curación de las secuencias se llevó a cabo visualmente en el programa Sequencher 4.1.4 (Arbor, 2010). Se obtuvo el marco de lectura del gen para cada secuencia en la plataforma en línea ORFfinder (Rombel, Sykes, Rayner, & Johnston, 2002). Posteriormente, las secuencias fueron sometidas en la base de datos genéticos Genbank (Benson et al., 2013) y utilizando la plataforma BLAST (Johnson et al., 2008) se compararon con registros previos para comprobar su identidad. Se construyó una base de datos con la información genética disponible en Genbank (Benson et al., 2013) y con registros nuevos que representan información genética para la Isla Gorgona y el Pacífico colombiano.
Diversidad genética: Los análisis se realizaron partir de un alineamiento de 414 pb entre las secuencias seleccionadas para este estudio. La diversidad nucleotídica (π) y el estadístico D de Tajima (Tajima, 1989) se estimaron en el programa Arlequin 3.5.2.2 (Excoffier & Lischer, 2010). La riqueza haplotípica se calculó a partir de un criterio 99.999 % de identidad nucleotídica entre todo el conjunto de secuencias COI siguiendo una metodología descrita previamente para malaria aviar (Gil & Sedano, 2019) y utilizando para esto el programa Usearch (Edgar, 2010).
Estructura genética: La estructura genética de A. cephalotes se estudió a escala continental (América) y a escala local (POT) utilizando la ubicación geográfica reportada para cada secuencia (Apéndice Digital 1). Se realizó un análisis de varianza molecular (AMOVA) del marcador COI en el programa Arlequin 3.5.2.2 (Excoffier & Lischer, 2010) entre las secuencias encontradas en la región trans-andina y aquellas de la región cis-andina. Adicionalmente, para la región del POT se construyó una red de haplogrupos utilizando el método “Median-Joining” en el programa PopArt (Röhl, Bandelt, & Forster, 1999) y se realizó una AMOVA entre las secuencias de los haplogrupos identificados en la isla con aquellas de los haplogrupos en el área continental del POT (Apéndice Digital 2).
Un análisis espacial de la estructura genética se desarrolló mediante una prueba de asignación de individuos para estimar el número probable de poblaciones que conforma la distribución de A. cephalotes. Con la incorporación de coordenadas geográficas en el análisis se considera de manera explícita la incertidumbre de la ubicación espacial de cada individuo y se puede estimar el número más probable de agrupaciones genéticas (k) alcanzando equilibrio Hardy-Weinberg (Rodriguez, 2013). Para esto se utilizó el paquete Geneland 4.0.8 del programa R 3.4 que implementa un algoritmo basado en un modelo Bayesiano y el método Monte Carlo de cadenas de Markov (Guillot, Estoup, Mortier, & Cosson, 2005). Este análisis confiere a cada individuo una probabilidad posterior de ser asignado en una agrupación k sin un conocimiento a priori sobre las unidades poblacionales o de sus límites geográficos (Guillot et al., 2005).
El modelo espacial asume que la dispersión para el género Atta es de 2 a 50 km (Solomon et al., 2008), se calculó grosso modo una capacidad máxima de dispersión media equivalente a 676 km2 y se realizó un análisis en Geneland (Guillot et al., 2005) con k = 6 como el número máximo de poblaciones. El análisis se repitió independientemente diez veces y se recuperaron 25 000 datos a partir de 1 000 000 de iteraciones en cada repetición. Para elegir la repetición que mejor se ajusta al análisis con un modelo espacial implementado en Geneland (Guillot et al., 2005) se utilizó un criterio de decisión a partir del logaritmo de los Factores de Bayes (LBF). Si al comparar dos modelos (modelo uno y modelo dos), el valor LBF < -2 entonces es preferible seleccionar el modelo dos, mientras que si LBF > 2 será preferible seleccionar el modelo uno (Kass & Raftery, 1995).
Se realizó la delimitación geográfica en el POT de una población de A. cephalotes, examinando la distribución de probabilidad que resulta de la asignación de individuos en Geneland (Guillot et al., 2005). La distribución de probabilidad es incorporada a un análisis complementario de conectividad que se fundamenta en la teoría de circuitos eléctricos y es desarrollado en el programa Circuitscape 4.0 (McRae, Shah, Mohapatra, & Anantharaman, 2014). En este análisis las características del paisaje de probabilidad se presentan en términos de conductividad o resistencia. Sobre este paisaje se itera una serie de caminatas aleatorias entre nodos que siguen un modelo Markoviano (McRae, Shah, & Mohapatra, 2013). El análisis de conectividad se utiliza para proyectar en un mapa el cálculo de la conductividad de la variación genética en términos del paisaje de probabilidad de asignación de individuos. Altos valores de conductividad indican mayor conectividad entre nodos, y en este caso, los nodos corresponden a cada uno de los sitios de muestreo. La variación de la conectividad entre dos nodos representa en cierto modo un cambio espacial en la variabilidad genética. Además, en este análisis la cordillera de los Andes se constituye a priori como una barrera asumiendo un límite altitudinal por encima de los 1 500 m.s.n.m para la distribución de A. cephalotes entre la región trans-andina y cis-andina.
Análisis demográfico: Se realizó un análisis de coalescencia para examinar modelos de migración de A. cephalotes entre el área continental y la Isla Gorgona con el programa Migrate 4.2.14 (Beerli, 2017) y posteriormente estimar el tamaño efectivo poblacional (θ) y la tasa de migración (M) escalados a la tasa de mutación utilizando el modelo que mejor se ajusta a los datos. Mediante un análisis bayesiano (Beerli, 2016) este programa utiliza información de la genealogía, considera la incertidumbre en la estimación propia de la genealogía y asume θ constante, o que a lo sumo varía aleatoriamente alrededor de su valor medio, resultando más informativo que el índice de fijación F st calculado con un AMOVA.
En Migrate se programó una cadena de 900 000 genealogías, se descartaron 300 000 árboles iniciales y con 20 repeticiones independientes se hizo un promedio para la estimación de los parámetros. Para esto se utilizó un modelo de substitución HKY ajustado (igual frecuencia para todas las bases), seleccionado en el programa jModelTest 2.1 (Darriba, Taboada, Doallo, & Posada, 2012; Guindon & Gascuel, 2003), y una proporción Ti/Tv = 2.0 para estimar las frecuencias nucleotídicas de los datos. Se implementó el modo adaptativo de cadenas calientes, donde en cada paso se realizan intercambios de aceptación y rechazo en las estimaciones de los parámetros, para asegurar una búsqueda independiente y comprensiva del espacio estadístico de los parámetros. La estacionalidad de la simulación fue determinada por altos valores ESS de cada parámetro. La integración termodinámica de la aproximación de Beziér (implementada en Migrate) fue utilizada para estimar el valor marginal de verosimilitud y calcular la probabilidad condicionada a los datos de los diferentes modelos. Con los modelos poblacionales de mayor probabilidad se reinterpretan los valores del parámetro M como una aproximación al número de inmigrantes efectivos por generación (Nm) (Beerli, 2016), utilizando el programa R 3.4.4 (R Core Team, 2017).
Filogenia y tiempo de divergencia: Una reconstrucción de la genealogía para los haplogrupos de A. cephalotes se realizó en el programa Beast 1.8.4 (Suchard et al., 2018), disponible en Cipres (Miller, Pfeiffer, & Schwartz, 2010). El análisis se implementó considerando un reloj molecular relajado con distribución Lognormal y un modelo HKY ajustado para la substitución nucleotídica (igual frecuencia para todas las bases). Además, se propuso un tamaño constante para la población y se asignó una distribución normal para la tasa de substitución con un valor medio de 9.5 substituciones por sitio por millón de años (Solomon, 2008).
Resultados
La base de datos para el gen mitocondrial COI de A. cephalotes contiene un total de 422 secuencias de tamaño variable (224-721 pb) reportadas para 14 países de América (Apéndice Digital 3). Esta compilación de secuencias corresponde a 389 registros de GenBank (Benson et al., 2013) y 33 registros secuenciados de novo para la región del Pacífico colombiano (Apéndice Digital 1) con un marco de lectura abierto de 414 pb (MG948859 - MG948889, MG948891, MG948895).
Se identificaron 42 grupos haplotípicos en total, entre las 422 secuencias de la base de datos. En el POT se encuentran 18 haplogrupos, 16 de ellos son endémicos de esta región (Apéndice Digital 2). Los valores estimados para el estadístico D de Tajima en cada localidad sugieren que el marcador COI satisface la condición de neutralidad y los valores negativos observados en la mayoría de las agrupaciones indican un exceso de alelos pocos comunes en el POT (Tabla 1).
Diversidad de haplogrupos del gen COI de A. cephalotes entre localidades de la región trans-andina y la región cis-andina como un único grupo. de la prueba de neutralidad Diversity of COI gene haplogroups of A. cephalotes for trans-andean locations and the cis-andean region as a whole
TABLA 1Diversidad de haplogrupos del gen COI de A. cephalotes entre localidades de la región trans-andina y la región cis-andina como un único grupo. de la prueba de neutralidadTABLE 1Diversity of COI gene haplogroups of A. cephalotes for trans-andean locations and the cis-andean region as a whole | ||||||||||
| Localidades | N | H | Hd | π | Tajima D | P-valor | ||||
| Andes (Colombia) | 7 | 3 | 0.0545 | 0.5238 ± 0.2086 | -1.2371 | 0.1179 | ||||
| Buenaventura | 2 | 2 | 0.0363 | 1.0000 ± 0.5000 | 0.0000 | 1.0000 | ||||
| Dagua | 7 | 4 | 0.0727 | 0.7143 ± 0.1809 | -1.4341 | 0.0629 | ||||
| Gorgona | 10 | 2 | 0.0363 | 0.3556 ± 0.1591 | 0.0149 | 0.7352 | ||||
| Guapi | 7 | 4 | 0.0727 | 0.8571 ± 0.1023 | -0.8764 | 0.2721 | ||||
| Quibdó | 7 | 4 | 0.0727 | 0.7143 ± 0.1809 | -0.6540 | 0.3223 | ||||
| Costa Rica | 14 | 4 | 0.0727 | 0.6484 ± 0.1163 | -0.2600 | 0.4437 | ||||
| Ecuador | 8 | 3 | 0.0545 | 0.4643 ± 0.2000 | -0.4479 | 0.3318 | ||||
| Panamá | 9 | 6 | 0.1090 | 0.8889 ± 0.0910 | -1.0690 | 0.1860 | ||||
| cis-Andes | 64 | 23 | 0.4181 | 0.8036 ± 0.0509 | 0.1428 | 0.6288 | ||||
N: número de muestras, H: número de haplogrupos, Hd: diversidad haplotípica, π: diversidad nucleotídica ± desviación estándar, el índice D de Tajima con su valor-P.
N: sample size, H: haplotype groups, Hd: haplotypic diversity, π: nucleotidic diversity and the standard deviation, Tajima’s D and neutrality test P-value.
Análisis poblacional a escala regional: El análisis de asignación de individuos implementado en Geneland (Guillot et al., 2005) indica k = 4 poblaciones como el número más probable de agrupaciones genéticas que conforma A. cephalotes (LBF ≤ -176.2542). Este resultado constituye una primera aproximación a la delimitación geográfica de la estructura genética para A. cephalotes en el POT (Apéndice Digital 4). El análisis de conectividad implementado en Circuitscape (McRae et al., 2013) facilitó definir límites geográficos más precisos para una población que coincide espacialmente con la región Dominio Pacífico según la caracterización de regiones biogeográficas (Morrone, 2014). Esta población de A. cephalotes se denomina de ahora en adelante Dominio del Pacífico y se distribuye en Costa Rica, Panamá, la mayor parte del occidente de Colombia y el norte trans-andino del Ecuador (Fig. 1 y Apéndice Digital 4.A).
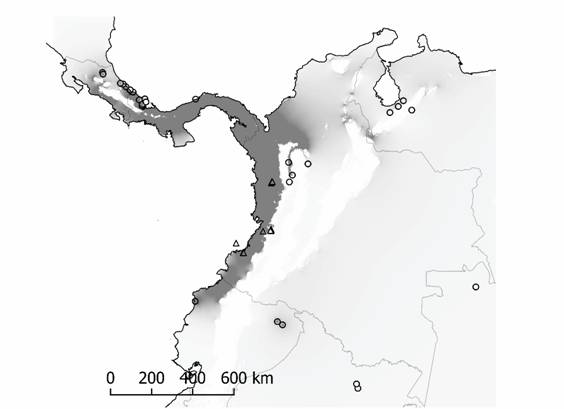
Fig. 1. Fig. 1 Conectividad de la variabilidad genética de A. cephalotes en el Pacífico Oriental Tropical. A mayor conectividad espacial, más oscuro es el sombreado de gris en el mapa, y viceversa. La estimación de conectividad usa la interpolación de la probabilidad de asignación de individuos del análisis en Geneland (ver métodos y Apéndice Digital 4.A). Las accesiones del GenBank (círculos) y las muestras de novo para este estudio (triángulos) (ver Apéndice Digital 1). Spatial connectivity of the genetic variability for A. cephalotes in the Eastern Tropical Pacific. A higher connectivity across space is shown in darker gray-shade on the map, and vice versa. The connectivity estimates use spatial interpolation of probabilities from the individual assignment test using Geneland (see methods and Digital Appendix 4.A). GenBank accessions (circles) and de novo samples for this study (triangles) (see Digital Appendix 1).
El hecho que la mayoría de grupos haplotípicos identificados en la población Dominio del Pacífico sean en su mayoría endémicos de la región trans-andina es consistente con la alta probabilidad de asignación de individuos en esta población (Apéndice Digital 4.A). Existe un patrón de distribución diferencial de la variación genética de A. cephalotes entre las regiones trans-andina y cis-andina (AMOVA Φst 1 = 0.2749, Prueba Exacta de Distribución Individual P = 0.0000001). En la región trans-andina la diversidad genética de A. cephalotes es mayor que aquella de la región cis-andina (Tabla 1).
Análisis poblacional a escala local: En la Isla Gorgona los individuos de A. cephalotes son genéticamente similares a los individuos de localidades cercanas en áreas continentales de Colombia y en contraste, menos parecidos a las hormigas cortadoras del área continental en el Ecuador o en América central (Tabla 2). En Gorgona están representados sólo dos grupos haplotípicos de A. cephalotes, H16 y H17. Ambos son frecuentes en el Dominio del Pacífico con una distribución discontinua en la zona continental sin que medie una barrera geográfica aparente (Fig. 2A). La distribución espacial de H16 y H17 sugiere la delimitación latitudinal (5°6’ N) de dos áreas en el margen continental del POT en Colombia. Estas áreas corresponden a una región hacia el sur que incluye las localidades de Quibdó, Dagua, Buenaventura, Guapi e Isla Gorgona, en el Pacífico colombiano y termina en su punto más al sur en Ecuador (0°4’ N). Otra región hacia el norte que incluye áreas hacia el norte de los Andes colombianos, Panamá y termina con su punto más al norte en Costa Rica (10°4’ N) (Fig. 2A). La genealogía de A. cephalotes muestra un patrón politómico extenso (Apéndice Digital 5). El tiempo de divergencia de H16 y H17 podría ser muy reciente pero esta estimación debe ser interpretada cuidadosamente debido a que el nodo que relaciona H16 y H17 con su ancestro común más cercano presenta un bajo apoyo, 0.75 de probabilidad posterior (Apéndice Digital 5).
TABLA 2 TABLE 2 Distancia genética para gen COI de A. cephalotes entre la Isla Gorgona y áreas continentales del Pacífico Oriental Tropical Genetic distance of A. cephalotes for gen COI among the Gorgona Island and mainland areas of the Eastern Tropical Pacific
| Localidades | F st 1 |
| Costa Rica | 0.42374* |
| Panamá | 0.26829* |
| Andes (Colombia) | 0.05405 |
| Quibdó | 0.17586 |
| Buenaventura | 0.68750 |
| Dagua | 0.61827* |
| Guapi | 0.12773 |
| Ecuador | 0.91734* |
1 Índice de fijación entre grupos.
* Diferencias P < 0.05.
Existe evidencia de diferenciación genética dentro del área continental como también entre la Isla Gorgona y las áreas continentales del POT (Fig. 2A). Se encontró diferenciación de A. cephalotes entre la Isla Gorgona y la región sur de la costa pacífica (AMOVA Φst 2 = 0.1641, Prueba Exacta de Distribución Individual P = 0.01366) (Fig. 2A). Igualmente existe diferenciación genética entre la Isla Gorgona y la región norte de la costa pacífica (AMOVA Φst 2 = 0.1828, Prueba Exacta de Distribución Individual P = 0.00545) (Fig. 2A). Es informativo señalar que también existe diferenciación genética entre las regiones norte y sur dentro del área continental (AMOVA Φst 2 = 0.22269, Prueba Exacta de Distribución Individual P = 0.00001).
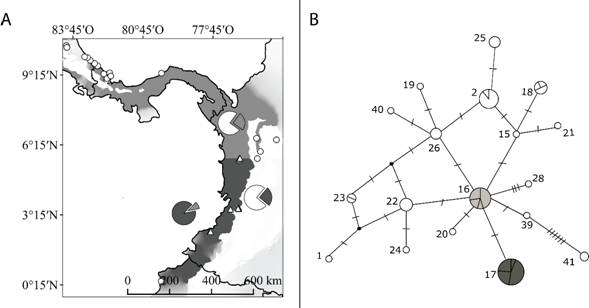
Fig. 2. Fig. 2 (A) Distribución de los haplogrupos H16 (gris claro) y H17 (gris oscuro) y la frecuencia de estos haplogrupos en el área continental y en la Isla Gorgona (gráfico circular). (B) Red de haplogrupos en el Dominio del Pacífico. El tamaño de los círculos es proporcional a la frecuencia de cada haplogrupo, a mayor tamaño mayor número de secuencias en la muestra. En la red cada barra entre los círculos equivale a un paso mutacional y los puntos negros representan un ancestro hipotético. Los círculos en color gris claro y gris oscuro representan los haplogrupos H16 y H17, respectivamente; y los círculos blancos representan la proporción de los otros haplogrupos numerados en el Dominio del Pacífico (Apéndice Digital 2). (A) Distribution of haplotypic groups H16 (light grey) and H17 (dark grey) and the frequency of these haplogroups in the mainland and the Gorgona Island (pie charts). (B) Haplogroup network in the Pacific Domain. The size of the circles is proportional to the frequency of each haplogroup, that is, the larger the circle, the greater number of sequences in the sample. In the network, each bar between circles is equivalent to a mutational step and the black dot represent an unknown ancestor. The circles in light gray and dark gray represent the haplogroups H16 and H17, respectively; and the circles in white show the proportion of the other haplogroups numbered in the Pacific Domain (Apéndice Digital 2).
Los resultados del análisis de la coalescencia sugieren que dos de los siete modelos poblacionales examinados en Migrate-n acumulan aproximadamente el 92 % de la probabilidad en la selección de modelos (Apéndice Digital 5). Ambos modelos coinciden en indicar un flujo genético asimétrico entre la Isla Gorgona y el área continental del Pacífico, pero describen relaciones de ancestaría diferentes entre las poblaciones examinadas aquí. Los modelos poblacionales con mayor probabilidad, ambos, sugieren que existe migración unidireccional desde la isla hacia el continente (Apéndice Digital 6). La estimación de parámetros demográficos con estos dos modelos resulta en valores modales para θ continente, θ Gorgona y M Gorgona→Continente alrededor de 0.01430, 0.00271 y 508.0, respectivamente. El cálculo de Nm sugiere que cerca de 4 inmigrantes por generación se mueven hacia la población del Pacífico continental desde la isla. Sólo la distribución posterior de M Gorgona→Continente muestra que una parte de esta curva podría no diferenciarse de cero y por esto su interpretación debe realizarse con precaución (Apéndice Digital 7). Colectivamente, el análisis sugiere que el flujo podría ser bajo o que la cantidad de variación entre las secuencias parciales de COI podría ser insuficiente para la estimación de una distribución de M con todos sus valores diferentes a cero.
Discusión
Las nuevas secuencias del gen COI de A. cephalotes de la Isla Gorgona y el Pacífico colombiano cubren una brecha de información en el POT que se suman a los aportes de previos estudios (Martins et al., 2007; Solomon et al., 2008; Tranter, 2016). Sabemos ahora que el Pacífico colombiano, incluyendo la Isla Gorgona, alberga una proporción importante de la diversidad genética de A. cephalotes; aproximadamente 23 % de la diversidad genética que ha sido caracterizada para esta especie en Suramérica. Así, una región que equivale a menos del 1 % del área global, acumula cerca de un cuarto de la diversidad genética de herencia matrilínea para esta especie.
De este modo fue posible hacer una aproximación espacial de la estructura genética en el POT para uno de los organismos más abundantes y con más amplia distribución en el continente americano. Los resultados de la asignación de individuos de A. cephalotes en cuatro agrupaciones espacialmente definidas en este estudio no coinciden con la asociación de agrupaciones geográficas presentado por Solomon et al. (2008), esta aproximación filogenética para definir agrupaciones geográficas es inconsistente con el soporte de la topología del árbol. Dichas agrupaciones no son reproducibles debido a un extenso patrón de politomías en la genealogía del marcador COI, es decir, un patrón generalizado de bajo soporte de nodos que se verificó con un análisis de inferencia filogenética realizado en nuestro estudio (Apéndice Digital 5).
En contraste con el estudio previo, nosotros presentamos una aproximación a la biogeografía de A. cephalotes implementando un análisis de asignación de individuos en poblaciones con un modelo espacial explícito que tiene en cuenta la variación genética y la capacidad de dispersión conocida para la especie. Este modelo es sujeto de análisis probabilísticos bayesianos reproducibles que además pueden ser consistentemente extrapolados a un contexto geográfico bien definido, utilizando una metodología con un marco teórico robusto (Guillot et al., 2005).
La evidencia sugiere que la Isla Gorgona mantiene en sus 26 km2 el 11 % de los haplogrupos del gen COI en el Dominio del Pacífico, la población de A. cephalotes genéticamente más diversa del continente. La distribución espacial de los haplogrupos identificados en el Dominio del Pacífico indica que la Isla Gorgona pudo ser colonizada por reinas procedentes de la costa pacífica colombiana con las cuales presenta mayor identidad de ADN de herencia matrilínea. Aunque la capacidad de dispersión de A. cephalotes podría ser suficiente para promover la colonización de la isla desde áreas continentales próximas, eventos de introducción antropogénica de estas hormigas cortadoras no pueden ser excluidos como una explicación parsimoniosa para la colonización de ambos haplotipos en la isla.
Probablemente la colonización de la Isla Gorgona se dió desde latitudes al norte de la localidad de los Andes (6°15’ N) como también desde poblaciones al suroccidente colombiano como la localidad de Guapi, un puerto de embarcación con influencia en la isla a tan solo 60 km de distancia (Fig. 2.A). Este resultado es congruente con estudios previos que indican, como entre los artrópodos, los linajes que colonizan islas provienen de las áreas continentales más próximas (Edwards & Thornton, 2001; Garb & Gillespie, 2006, 2009; Gillespie & Roderick, 2002; Lourenco & Flores, 1989; Sharma & Giribet, 2012). Por otra parte, estudios en la Isla Gorgona sobre la diversidad y el proceso de dispersión de las especies de árboles han especulado que el tamaño de la isla limita la disponibilidad de hábitats (Yockteng & Cavelier, 1998).
Independientemente del mecanismo de dispersión de A. cephalotes para colonizar la Isla Gorgona, la estimación de parámetros demográficos indica que el flujo genético de A. cephalotes entre la isla y el continente puede estar restringido. Es posible que la abundante población de A. cephalotes en un área insular pequeña, como es el caso de la Isla Gorgona, facilite la continua dispersión de algunas reinas hacia áreas continentales próximas y en cambio restrinja el establecimiento en la isla de reinas inmigrantes que portan los grupos haplotípicos menos frecuentes de las áreas continentales. Así, el tamaño de la Isla Gorgona puede estar restringiendo el número de individuos en las poblaciones locales y aumentando potencialmente las tasas de extirpación en la isla.
Bajo nuestro análisis, los individuos de A. cephalotes que habitan las selvas del Pacífico colombiano, incluyendo la Isla Gorgona, conforma una población de hormigas cortadoras denominada aquí como Dominio del Pacífico. El rango de distribución geográfica de esta población puede ser acotada y representada en un mapa de conectividad genética, donde se observa que hacia los límites geográficos de la población es razonable predecir un cambio en su patrón de variación genética (Fig. 1 y Apéndice Digital 4A). Lo anterior permite especular que la diversidad genética de la región cis-andina colombiana conforma una agrupación divergente de aquella identificada en el POT. En estas fronteras sería interesante conocer cómo es la relación de la diversidad genética del Dominio del Pacífico con aquella de la región cis-andina de Colombia, Venezuela y Ecuador en una escala espacial más detallada.

