











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Las talasemias son alteraciones hereditarias que repercuten en la síntesis de globina sin afectar al grupo heme y se caracterizan por la producción disminuida de una cadena particular de globina y acumulación de otras cadenas de globinas no afectadas. Sus manifestaciones clínicas son variadas, desde anemias leves que pueden pasar desapercibidas, hasta padecimientos graves dependientes de transfusiones. Una característica común a los síndromes talasémicos (desorden autosómico recesivo con una expresión clínica variable según el tipo de mutación y del número de factores moduladores) es un cuadro hemolítico, con microcitosis hipocrómica y eritropoyesis ineficaz. Las talasemias se encuentran entre las enfermedades genéticas de mayor prevalencia. A la fecha, se han descrito más de 600 defectos moleculares en las beta-talasemias que reducen (β+) o no permiten que se produzca (β0) la expresión de cadenas de beta-globina.1,2
En términos generales, entre un 80% y 90% de los casos concentrados en un área geográfica, prevalece un número de mutaciones específicas comunes a esa región en particular. Sin embargo, mutaciones más raras, o menos comunes, también son detectadas en dichas áreas. Así ha sucedido también en Costa Rica, donde defectos poco frecuentes en relación con su zona geográfica han sido recientemente descritos.3, 4 Se describe aquí la identificación de una mutación común en el codón 39 (C>T) de la cadena de beta-globina y una deleción delta-beta (δβ0) siciliana 13.4 kb, cuya presentación como doble heterocigota no ha sido descrita anteriormente en la literatura científica y cuya presentación clínica causa una beta-talasemia (β0) transfusión dependiente.
Presentación de los casos
La probando 1 fue una paciente de 13 años 2 meses de edad, vecina de Limón, con historia familiar de madre portadora de delta-beta (δβ) talasemia. Desde los 4 años, la niña recibe seguimiento en la consulta externa del servicio de hematología del Hospital Nacional de Niños (HNN) Dr. Carlos Sáenz Herrera, a donde fue referida del Hospital Tony Facio, de Limón, luego de un internamiento por cuadro respiratorio. Al momento del internamiento, la paciente presentó un antecedente personal de anemia hemolítica con ambas pruebas de Coombs (directa e indirecta) negativas.
Durante su internamiento se realizaron serologías por Mycoplasma pneumoniae y gota gruesa, las cuales resultaron negativas. Además, se efectuó una curva de fragilidad osmótica (CFO) que reportó un desplazamiento a la izquierda de la curva, no compatible con esferocitosis hereditaria (EH). Asimismo, se llevó a cabo una electroforesis de hemoglobina, la cual se consignó en el expediente médico de la paciente como compatible con deltabeta (δβ) talasemia (Cuadro).

La paciente, desde septiembre de 2014 y hasta marzo 2020, recibió transfusión de glóbulos rojos cada 3 o 4 semanas con el fin de mantener un nivel de hemoglobina superior a 9 g/dL; sin embargo, presentó deformidad ósea, que era el objetivo por evitar con ese esquema transfusional, por lo que se intentó mantener el nivel superior a 7 g/dL. En el año 2011 se le realizó un ecocardiograma que evidenció un corazón de estructura y forma normal, con ventrículo izquierdo dilatado y fracción de acortamiento del 32%, típico de anemia crónica. Clínicamente, la niña ha mantenido requerimientos transfusionales como en un paciente con síndrome talasémico de beta-talasemia mayor, lo que no es concordante con el diagnóstico electroforético de delta-beta (δβ) talasemia. Por lo anterior, se realizó un estudio electroforético del padre, se documenta compatibilidad con heterocigoto para beta (β) talasemia. Lo anterior motivó a efectuar un estudio molecular de la probando 1 en el que se obtuvo un resultado de doble heterocigoto para una mutación común en el codón 39 (C>T), causante de beta-(cero)-talasemia, y una deleción (δβ0) siciliana 13.4kb, causante de delta-beta (cero) talasemia.
Durante el periodo del 3 de diciembre de 2010 al 26 de junio de 2020, la niña recibió 70 transfusiones, según registro del banco de sangre del HNN. La paciente requirió esplenectomía en julio de 2011 y colecistectomía en julio de 2018. Por complicaciones relacionadas con las transfusiones de glóbulos rojos, presentó hiperferritinemia por lo que recibió tratamiento con el quelante de hierro deferasirox. El 26 de junio de 2020, se le trasladó del hospital pediátrico a un hospital de adultos, por edad, de acuerdo con los lineamientos de la Caja Costarricense del Seguro Social (CCSS) de Costa Rica, en donde actualmente se encuentra en control y seguimiento sin otras complicaciones recientes.
La probando 2, de 14 años 2 meses de edad, quien no tiene relación alguna con la probando 1, inició control en el Servicio de Hematología del HNN desde los 3 años. Debutó clínicamente como paciente con anemia hemolítica Coombs negativa. En estudios realizados al inicio, se obtuvo una curva de fragilidad osmótica (CFO) con desviación a la izquierda, lo que descarta esferocitosis hereditaria. Se les efectuó un estudio electroforético a los padres, siendo la madre portadora de delta-beta (δβ) talasemia y el padre se documentó heterocigoto para beta (β) talasemia. Primeramente, se cataloga como homocigota para delta-beta-talasemia, lo que motivó a realizar un estudio molecular de la probando 2 con el que se logró documentar un estado de doble heterocigoto: una mutación común en el codón 39 (C>T), causante de beta-(cero)-talasemia (β0), y una deleción tipo siciliana 13.4 kb, causante de delta-beta (cero) talasemia δβ0.
La probando 2 se mantuvo con requerimientos transfusionales altos, con transfusiones de glóbulos rojos empacados cada 3 a 4 semanas con el objetivo de mantener un nivel de hemoglobina superior a 9 g/dL. A continuación, se cambió la meta a mantener niveles mínimos de hemoglobina en 7g/dL y fueron requeridas trasfusiones cada 6-8 semanas. Fue esplenectomizada en julio del año 2011 y colecistectomizada en julio del año 2018. Recientemente fue trasladada a un hospital de adultos. Al presente, está en seguimiento hematológico cada 6-8 semanas con requerimientos de transfusión de glóbulos rojos empacados con la misma frecuencia y recibe terapia quelante de hierro con deferasirox.
La sangre periférica de las pacientes se analizó en un equipo Sysmex XN1000 y se obtuvieron índices de serie roja compatibles con talasemia. Se realizó una electroforesis de zona capilar (CZE, por sus siglas en inglés) en equipo Capillarys II (Sebia Park Technology, Lisses, Francia). Los resultados obtenidos se observan en el Cuadro. Para el estudio molecular, se extrajo ADN genómico de los leucocitos de sangre periférica de ambas probandos. Se efectuó un estudio de secuenciación por Sanger y MLPA (por sus siglas en inglés, de multiplex ligation dependent probe amplification), para buscar mutaciones y deleciones en el grupo de genes que codifican para la cadena de beta-globina. El MLPA es un método comparativo basado en la amplificación cuantitativa y subsecuente análisis de los fragmentos de múltiples sondas que se hibridizan a una región de interés. Debido a que la amplificación de las sondas solo puede darse si existe un ADN complementario en la muestra, este método permite mostrar el perfil genético de la variación en el número de copias en el genoma del paciente. Se utilizó el kit MLPA P102-D1 para HBB (beta-globina) de la casa MRC-Holland (Amsterdam, The Netherlands), que contiene 49 sondas diseñadas para detectar cambios en el número de copias en el locus para la cadena β de hemoglobina (cromosoma 11p15.4) y que abarcan desde 1 Mb corriente arriba de la región del locus control, hasta 10 kb corriente abajo del gen de la β globina. Los productos de amplificación se separaron empleando electroforesis capilar en el secuenciador ABI PRISM 3100 (Applied Biosystems, Foster City, CA). Se utilizó el Gene Mapper versión 3.7 (Applied Biosystems) para la determinación del tamaño; los datos obtenidos fueron analizados con el software Coffalyser (MRC-Holland). Se emplearon muestras de controles sanos normales como control de las reacciones de la MLPA. En ambas probandos se documenta una mutación en el codón 39 (C>T) (β0) en combinación con la deleción siciliana δβ0 13.4 kb (Figura).
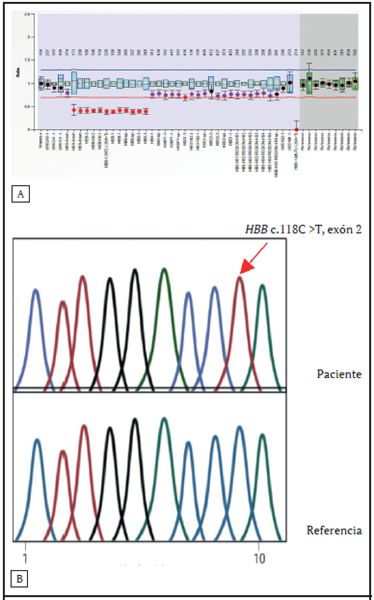
Figura 1. A. Representación alelo 1: se detectó una deleción en la región 11p15.4 de un tamaño aproximado de 8,1 kb (12 sondas que se extienden desde el exón 3 del gen HBD hasta la región corriente abajo del gen HBB, deleción reportada en la literatura como deltabeta 0 siciliana. B. Representación alelo 2: se detectó la variante c.118C>T, p.(Gln40Ter) ubicada en el exón 2 y en estado hemicigoto. Esta variante produce un codón de terminación prematuro y se asocia con B0 Talasemia.
Discusión
Según la tradición, las beta-talasemias se clasificaban clínicamente en tres categorías según la gravedad del cuadro: la talasemia mayor (βT / βT), las cuales son graves y dependientes de transfusiones; la talasemia menor, o con pocos síntomas, usualmente representada por el rasgo de portador (βA / βT) y la talasemia intermedia,5 la cual es extremadamente heterogénea, tanto respecto a su causa genotípica como a su fenotipo clínico, y va desde una anemia severa que requiere transfusiones sanguíneas intermitentes, a condiciones asintomáticas levemente mayores a las que presentan los portadores asintomáticos. En la actualidad, debido a la heterogeneidad clínica, la clasificación se ha dividido en dos categorías: talasemia dependiente de transfusión y talasemia no dependiente de transfusión; la primera presenta la mayor gravedad clínica.6 Uno de estos genotipos es el heterocigoto compuesto por un alelo β-tal leve o grave, cuyo resultado dependerá de si la mutación es de la variedad β+ o de la β0.
La condición heterocigota, por otra parte, tiene un cuadro fenotípico que semeja el rasgo de beta-talasemia con una HbA2 normal y una hemoglobina F (HbF) elevada que varía entre un 5% y 20%. Debido a que los homocigotos por deltabeta- talasemia tienen hallazgos idénticos por HPLC a los de homocigotos para persistencia hereditaria de HbF, los hallazgos clínicos de una anemia leve van más a favor de una delta-beta-talasemia y no de una persistencia hereditaria por hemoglobina fetal (PHHbF). Por ello, los estudios familiares juegan un papel crítico en el correcto diagnóstico. La deleción siciliana, por ejemplo, presenta una deleción de 13,379 pares de bases, remueve la región 3'del gen delta, la totalidad del gen beta y se extiende hasta 6.4 kb del elemento L1.7 Cuando las deleciones se combinan con mutaciones de beta-tal, justo y como en los dos casos descritos en este artículo, da como resultado fenotipos clínicos que abarcan desde condiciones asintomáticas hasta cuadros de beta-talasemia mayor. La sobreposición de los parámetros hematológicos y la variabilidad en la expresión de la HbA2 y Hb F hacen necesario el estudio molecular para la distinción de las deleciones y mutaciones clínicamente relevantes. Se describen aquí los primeros dos casos en Costa Rica debidos a una asociación de la mutación en el codón 39 (C>T) (β0) en combinación con la deleción siciliana δβ0 13.4 kb. La mutación en el codón 39 (C>T) ha sido descrita en 6.25% de los casos de beta-talasemia en Marruecos8 y con una frecuencia de 3.8% en los casos de beta-talasemia en Algeria. Sin embargo, esta es la primera vez que se detectan en nuestro país, la mutación puntual en el codón 39 (C>T), la cual es muy común en otros países, y que se presenta concomitantemente con la deleción δβ (0) tipo 13.4 kb siciliana. A la fecha no se ha encontrado reporte en la literatura científica de una doble heterocigosis semejante a los dos casos aquí descritos. En áreas donde la talasemia es prevalente, la detección de las mutaciones por delta-betatalasemia son muy importantes debido a que la coexistencia de esta con las alfa o beta-talasemias podría causar un diagnóstico erróneo o incluso un no diagnóstico por talasemia.9 Por lo tanto, estos casos recalcan la importancia del genotipeo de pacientes con delta-beta talasemia o PHHbF, el cual juega un papel crucial en la determinación de la fisiopatología y en el consejo genético. El laboratorio debe ser capaz de discriminar entre una hemoglobina fetal (HbF) elevada artefactualmente y otras causas clínicas serias, entre las que se encuentran: malignidades en médula ósea, anemia aplásica, beta-talasemia transfusión dependiente (antes llamada talasemia mayor), delta-beta talasemia o condiciones no patológicas inducidas por mutaciones o polimorfismos que ocurren en el promotor del gen gamma.10 La detección de trastornos de la hemoglobina por deleciones o mutaciones conocidas permite un adecuado manejo clínico y el correspondiente asesoramiento genético. Con estos casos se resalta la importancia entre la concordancia de la clínica del paciente y los resultados de laboratorio, ya que cuando no se presenta esa concordancia, es necesario seguir estudiando al paciente con el fin de establecer un diagnóstico certero.

