











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introduction
Trypanosoma cruzi, a causal agent of Chagas’ disease, is a parasite that presents alternation in its life cycle between an invertebrate (triatomine) and vertebrate (mammal) host. Individuals with Chagas’ disease may present cardiological, digestive, and neurological symptoms and might die. Currently, 6 to 7 million people are affected by the disease, and 25 million are at risk of infection (WHO, 2023). In Venezuela, increases in prevalence and oral transmission outbreaks have been described in several states (Alarcón de Noya et al.; 2015; Añez et al.; 2020).
The parasite response is essential for viability in the heat and osmotic stress during the transition from the insect to the mammalian host, as well as when invading mammalian cells. The contribution of heat shock proteins in morphological and functional changes during stress situations has been implicated. This class of proteins is divided into several conserved families according to molecular weight and broad function, among which the five major families are HSP104, HSP90, HSP70, chaperonins, and small HSPs (Folgueira & Requena, 2007; Urményi et al.; 2014). They participate in protein folding and cell signaling processes and play an important role in the host’s immune response. They have been identified as dominant antigens in diseases caused by protozoa (De Andrade et al.; 1992; Requena et al.; 1993; Angel et al.; 1996; Menezes-Souza et al.; 2014) and helminths (Rothstein et al.; 1989; Moser et al.; 1990; Colebrook & Lightowlers, 1997; Ferrer et al.; 2005).
There is evidence that indicates that in T. cruzi Hsp are constitutively expressed and induced by different types of stress.
In particular, 90 kDa heat shock proteins (HSP90) are involved in numerous intracellular processes in eukaryotic cells. Studies have shown that in T. cruzi HSP83 (homolog of HSP90) is essential for cell division and control of the response to thermal stress (Jhonson, 2012). The overexpression and the participation of HSP83 in the differentiation processes of T. cruzi have been suggested, and the inhibition of HSP83 of T. cruzi produces arrest in proliferation, demonstrating the importance in the control of the cell cycle (Graefe et al.; 2002).
To date, there are merely two old reports about cloning this molecule (Dragon et al.; 1987; Nadeau et al.; 1992), and only its genomic structure and ATPase activity have been studied. This study aimed to perform the cloning, bioinformatics characterization, and expression of the heat shock protein HSP83 of T. cruzi (EPm6 clon, MHOM/VE/2007/Elpidio Padrón 6c) coding gene. The study would enable subsequent studies on the adaptation processes of the parasite to different microenvironments, the identification of new therapeutic or diagnostic targets, and the study of host-parasite interaction in Chagas disease.
Methodology
Parasite Material
T. cruzi epimastigotes obtained from a human Chagas disease case (EPm6 clon, MHOM/VE/2007/Elpidio Padrón 6c) were cultivated at 27 ºC in liver infusion tryptose broth (LITB) (Contreras et al.; 1994). Parasites were counted employing a Neubauer chamber and phase contrast microscope (Nikkon, Optiphot). They were adjusted to 1.5 x 109 epimastigotes/mL. Subsequently, the parasites were pelleted by centrifugation at 8,000 rpm for 15 minutes at four °C (IEC Centrifuge, B22M) and washed twice in 0.15 M phosphate saline buffer (PBS) pH 7.2 (57 mM Na2HP04, 18 mM KH2P04, 76.9 mMNaCl) to eliminate the remains of the culture medium. Finally, they were stored at -80 ºC until their use.
RNA Extraction and the Complementary DNA (cDNA) Obtaining
RNA extraction was carried out through the FavorPrep™ Viral Nucleic Acid Extraction Kit (Favorgene), where the parasite masses were previously treated with lysis buffer supplemented with Proteinase K (0.45 mg/mL) and RNase (80 U/ µL). Then, the extraction continued with the instructions described in the commercial kit. The complementary DNA (cDNA) was obtained by implementing the Reverse Transcription-Polymerase Chain Reaction technique and using the Access RT-PCR System (Promega) commercial kit. Specific primers were designed manually using BioEdit® Software v7.1.11 (Hall, 2013) and, as a template sequence, the hsp83 of a reference strain of T. cruzi of 2115 bp (Trypanosoma cruzi strain CL Brener hsp83) obtained from GenBank (Accession No. XM_809799) strain used in the T. cruzi genome project (El-Sayed et al.; 2005). The primers used were TcHSP83-D (5’- ATGACCGAGACATTCGCA -3’) and TcHSP83-R (5’- CTAGTCAACCTGCTC-CAT -3’). The reaction mixture consisted of AMV/Tfl reaction buffer, deoxynucleotide triphosphates (0.2 mM), forward and reverse primers (1 µM) each, MgSO4 (1 mM), AMV Reverse Transcriptase (0.1 U /µL), Thermus flavus (Tfl) DNA polymerase (0.1 U / µL), 10 µL of the extracted RNA sample, resulting in a final reaction volume of 50 µL.
The amplification reaction was carried out in an automatic C1000™ Thermal Cycler (BioRad) using the amplification program. The reverse transcription was conducted at 45 °C x 45 min; the initial denaturation, at 94 °C x 2 minutes, 30 cycles (denaturation at 94 °C x 1 min, hybridization at 60 °C x 1 min, extension at 68 °C x 2 min); and the final extension, at 68 °C x 7 min.
Electrophoresis
The amplification products of RTPCR reactions were detected by electrophoresis (2% agarose gel, stained with 0.5 μg/ mL ethidium bromide). The DNA migration was conducted in a horizontal electrophoresis chamber (Minicell * EC 370M), with a constant 60-100 V voltage and TAE buffer (40 mM Tris-acetic acid, 0.5 M EDTA, pH 8). DNA bands were visualized on a photodocumentation system, Gel Doc 1000 (Bio-Rad®), using the Multi-Analyst Program (Bio-Rad®); the size of the amplification bands was compared with a molecular marker of 1 kb DNA Ladder (Bioneer).
cDNA Purification and Cloning
The cDNA samples to be purified were fractionated on agarose gels and prepared in the TAE buffer, and the bands were visualized on the ultraviolet light transilluminator. The fraction of interest was cut with a sterile scalpel, and the DNA was eluted with the commercial Wizard®SV Gel and PCR Clean-Up System kit (Promega), according to the manufacturer’s instructions. For the ligation reaction of the purified PCR products and the commercial vector pGEM®-T Easy, 1 U of the T4 DNA ligase enzyme was used. A plasmid-insert molar ratio 1: 5 was used in (30 mM Tris-HCl (pH 7.8), ten mM MgCl2, ten mM DTT, one mM ATP) buffer and incubated at four °C for 12 h. After confirming the hsp83 into pGEM®-T Easy, this recombinant molecule was digested, and the purified cDNA was subcloned in the expression plasmid pQE30 (Qiagen, Valencia, CA).
cDNA Sequencing and Bioinformatic Analysis
Sequencing of cloned cDNA was achieved at the Sequencing Service of the National Center for Microbiology, Instituto de Salud Carlos III, Madrid, Spain, using 373A system, Model 377, Applied Biosystem, following the protocol BigDye Terminator Cycle Sequencing Ready Reaction Kit (ABI-PRISM, PE Biosystems). The cDNA sequence obtained was compared with the sequences included in the databases (nucleic acid and proteins) (GenBank, EMBL) and analyzed by bioinformatics programs. Multiple alignments were done using the Clustal W. The alignment was revised using BioEdit Sequence Alignment Editor, version 7.0.5.3 (Hall, 2013). Nucleotide and amino acid sequences were analyzed using the EditSeq program of DNAstar (Lasergene®, Madison, USA). Identity and similarities were looked for in the databases (protein and nucleic acid) (EMBL, GenBank) by BLAST (Boratyn et al.; 2019). Other analyses were performed using CDD-Search of the NCBI (National Center for Biotechnology Information) (Lu et al.; 2020), Interpro of the EBI (European Bioinformatics Institute) (Mitchell et al.; 2019), Motif scan, and ExPASy (Expert Protein Analysis System) of the SIB (Swiss Institute of Bioinformatics) (Proteomics Server) (Artimo et al.; 2012). Epitopes B prediction was performed using the Protean program from DNAstar (Lasergene®, Madison, USA). In addition, the program BcePred (Prediction of continuous B-cell epitope in antigenic sequences using physico-chemical properties) (Saha et al.; 2005) was employed.
cDNA Expression
Recombinant His-fusion proteins were expressed in Escherichia coli M15 (Qiagen, Valencia, CA) by induction with isopropyl-β-D-thiogalactopyranoside (IPTG, one mM) (Sigma, Madrid, Spain). The cultures were lysed using sonication and examined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the recombinant proteins were purified by affinity chromatography with nickel agarose as described by the manufacturer (Qiagen, Valencia, CA). The protein concentration was determined by the Bradford technique (Bradford, 1976). The purity of the proteins was analyzed by SDS polyacrylamide gel electrophoresis (SDS-PAGE), done under denaturing conditions according to Laemmli (1970), on 10% separation gel and 4.5% packing gel at 15 mA. After separating the sample, the gel was stained with Comassie Blue-Silver (De Moreno et al.; 1985) for 45 min, then placed in the bleaching solution until the sharp bands were visible. The molecular weight (MW) of the expressed proteins was compared with the Broad Range Protein Molecular Weigh Markers (Promega).
Western Blot (Immunoblotting)
After the proteins were separated by electrophoresis, they were transferred to a nitrocellulose membrane, according to Towbin et al. (1979), using a wet system and the Mini Trans-Blot® Cell (BioRad) equipment. For this, two sponges and two filter papers the size of the gel, impregnated in transfer buffer and the nitrocellulose membrane, were placed, and the gel was placed on the membrane. Then, two filter papers and a sponge equally impregnated with the buffer were placed in the same way and transferred at 100 mA for one hour at four °C. After the transfer had elapsed, the nitrocellulose membrane was incubated in blocking solution (0.15M PBS pH 7.2, 7.5% skim milk) overnight at four °C. Subsequently, it was incubated with anti-Epi rabbit hyperimmune serum (against T. cruzi epimastigotes) in a 1/1500 dilution in reaction solution (0.15 M PBS pH 7.2, 150 mM NaCl, ten mM Tris-HCl pH 7.4) for 90 min at 37 °C. Then, eight washes of 5 min each were performed with the wash solution (0.15 M PBS pH 7.2, 0.1% Tween 20) and subsequently incubated with the conjugate, anti-rabbit IgG coupled to peroxidase at a dilution of 1/3000 in reaction solution (PBS 0.15 M pH 7.2, NaCl 150 mM, Tris-HCl 10 mM pH 7.4) and under the same conditions described above; then, the washes were repeated. The visualization of the immune complexes was carried out by chemiluminescence incubating the membrane with the mixture provided by the commercial Super Signal® West Pico kit (Pierce) for one minute in the dark and developing using a photographic plate according to the manufacturer’s instructions with an exposure time of three minutes.
Analysis and Results
By RT-PCR, the cDNA was obtained with an approximate size of 2000 bp, which corresponds approximately to the expected size of the hsp83 molecule of T. cruzi.Figure 1A shows the bands obtained by RT-PCR from the RNA samples extracted with the different commercial kits. For gene expression, M15 transformed cells were induced with IPTG, control aliquots were taken from uninduced cells and post-1, 2, and 3 hours of induction, and analyzed on Coomassie-Silver stained acrylamide gel. The expected size of the recombinant protein, according to the deduced amino acid sequence, is 80.7 kDa + 1 kDa of the histidine tail; so, a molecular weight of almost 83 kDa would be expected. Figure 1B shows the discrete expression of a protein of nearly 83 kDa in sample two at the second hour of incubation and slightly higher at the third hour. Additionally, bands that are not present in the uninduced control are observed and appear at the second and third hour of post-induction (Figure 1B); perhaps they are truncated forms of the protein, which the bacteria tend to eliminate.
The recombinant protein was purified by affinity chromatography from the cultures induced at 3 hours with IPTG. Each fraction obtained by chromatography and the sediment and the supernatant before and after passing through the Ni-NTA nickel column were analyzed using a polyacrylamide gel. For the identification of the recombinant protein, the fractions obtained from the purification of the recombinant protein HSP83 from T. cruzi were electrophoresis (SDS-PAGE 10%) and, subsequently, electroblotting on a nitrocellulose membrane. The nitrocellulose membrane was incubated with hyperimmune rabbit serum anti-Epimastigotes of T. cruzi; after development with luminol, a specific reaction with the recombinant protein is evidenced (Figure 1C).
purified recombinant protein.
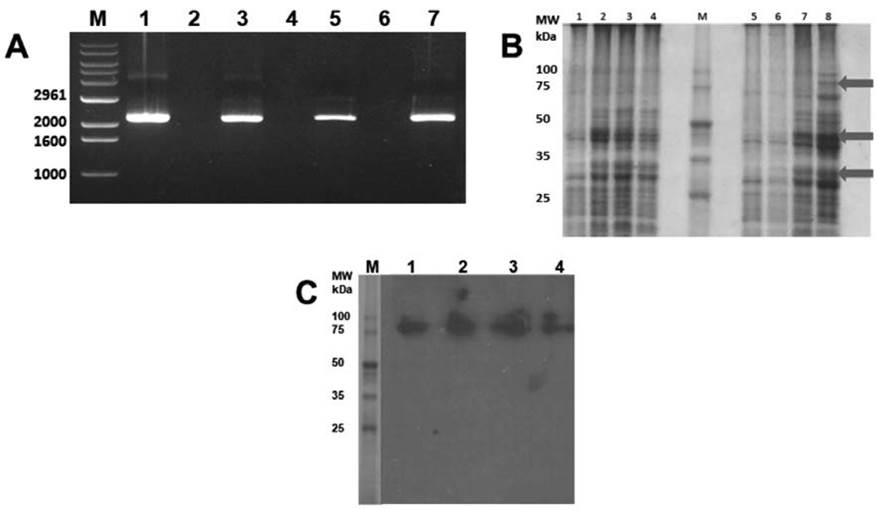
Figure 1 Cloning and expression of HSP83 of T. cruzi. A) Bands obtained by RT-PCR from the RNA samples. B) Transformed M15 cells induced with IPTG (1, 5), control aliquots taken from uninduced cells and post-1, 2, and 3 hours of induction (2, 3, 4 and 6, 7, 8, respectively), and analyzed on Coomassie-Silver stained acrylamide gel. C) Immunoblotting using hyperimmune rabbit serum anti-Epimastigotes of T. cruzi to detect purified recombinant protein.
The complete sequence of hsp83 cDNA was a fragment of 2115 bp corresponding to the open reading frame (ORF). This frame coded for a peptide of 704 amino acids, with a molecular mass of nearly 83 kDa and an isoelectric point of 5.0. The deduced amino acid sequence showed an amidation site (10-13), four possible N-glycosylation sites (48-51, 116-119, 151-154, 452-455), three cAMP and cGMP-dependent protein kinase phosphorylation sites (246249, 273-276, 329-332), 11 Casein kinase II phosphorylation sites (104-107, 155-158, 164-167, 291-294, 296-299, 306-309, 360363, 525-528, 531-534, 609-612, 621-624), four N-myristoylation sites (160-164, 190195, 232-237,442-447), nine Protein kinase C phosphorylation sites (104-106, 291-293, 332-334, 380-382, 410-412, 531-533, 553555, 644-646, 685-687), and four Tyrosine kinase phosphorylation sites (57-65, 218225, 273-281, 616-623). A motif and several domains were found, such as a Chaperone protein htpG (htpG) motif (80-704), a Heat shock hsp90 proteins family signature (103-112), a TonB-dependent receptor proteins signature 1 (1-40), two Bipartite nuclear localization signals (400-414, 554571), a Glutamic acid-rich region profile (257-327), a HATPase_cHistidine kinase-, DNA gyrase B-, a HSP90-like ATPase domain (105-258), and a HSP90 protein domain (261-704) (InterPro IPR001404 pFam PF00183). With respect to the possible immunogenicity of the complete sequence of hsp83 cDNA, five B epitopes were predicted in the molecule (226TKNEE230, 244EEGEKKKKT252, 327FEPSKK332, 470TGDSKK475, and 527EETEEEKKQR536) (Figure 2).
This sequence showed high identity with heat shock protein 85 (Trypanosoma cruzi Dm28c, Venezuela) (ESS64949.1), with Heat shock-like 85 kDa protein (Trypanosoma cruzi Dracon, Perú) (P06660.1) and lower identity with heat shock protein 85, putative (Trypanosoma cruzi CL Brener El Sayed, Brazil) (XP_811791.1) (Figure 3).
Discussion
The cloning of genes and their expression as recombinant proteins facilitates the

Figure 2 Schematic representation of the cloned cDNAs analyzed. The bars below deduced proteins represent the domains predicted in the sequence. The short grey lines below numbers represent the positions of the B epitopes; bp stands for base par, and aa means amino acid.

Figure 3 Clustal Alignment with heat shock protein 85 (Trypanosoma cruzi Dm28c, Venezuela) (ESS64949.1), Heat shock-like 85 kDa protein (Trypanosoma cruzi Dracon, Perú) (P06660.1), and heat shock protein 85, putative (Trypanosoma cruzi CL Brener El Sayed, Brazil) (XP_811791.1).
study of both the genes and the proteins they encode, as well as their use as tools in tests that help to elucidate the cellular mechanisms in which they are involved. In the present study, it was possible to clone and express the gene encoding the heat shock protein HSP83 from T. cruzi. To date, there are merely two old reports about cloning this molecule (Dragon et al.; 1987; Nadeau et al.; 1992), and only its genomic structure and ATPase activity have been studied. In addition, cloning and expression of the gene encoding HSP83 has been obtained in other tripanosomatids, such as Trypanosoma brucei (Mottram et al.; 1989), Hsp83 from Leishmania mexicana amazonensis (Shapira & Pinelli, 1989), Hsp83 of Leishmania infantum (Angel et al.; 1996), and Hsp83 from Leishmania braziliensis(Silva et al.; 2013; Menezes-Souza et al.; 2014).
Since HSP83 has been reported to be a protein highly expressed by T. cruzi, even under basal conditions (Dragon et al.; 1987), and given its importance in the mechanisms of cellular metabolism of the parasite and the need to expand studies in this area, it was decided to address the cloning strategy using the mRNA as a starting sample and the amplification of the coding region by means of the RT-PCR technique. Thanks to the information from the T. cruzi Genome project, this sequence was made available to design the cloning strategy.
In all cases, a band with a size of 2139 bp was amplified, the expected size according to the GenBank sequence. The verification of the cloning by colony PCR, using the primers for the coding region of HSP83, helped amplification of a band of 2139 bp in eight of the 19 colonies analyzed. The lack of expression in a high percentage of colonies analyzed could be due to the conditions of the cells used in cloning (which were not as competent at accepting the plasmid with the gene).
Likewise, the cDNA was subcloned into different expression vectors to facilitate its expression. However, despite successful verification of the insert’s presence in the recent subcloning, the HSP83 sequence or the entire plasmid disappeared in some cases when trying to express it. This phenomenon suggests that the HSP83-plasmid construct may be unstable within these complex plasmids, and bacteria may degrade the plasmid (with the loss of the HSP83 sequence) or even eliminate it (Maizels et al.; 1991).
Subsequently, it was decided to carry out the subcloning process in the vector pQE30, which was simpler and used for the cloning and expression of HSP83 from Leishmania infantum (Angel et al.; 1996). However, the same result occurred: subcloning was achieved but followed by loss. This reinforces the recurrence of the hypothesis that the recombinant plasmid is expelled from the bacterium, perhaps because the HSP83 sequence, being evolutionarily conserved, can interfere with the cell’s metabolism, and the cell tends to eliminate it (Huynh et al.; 1985). On the other hand, sometimes fragments of genetic material do not always integrate precisely; this may lead to artifacts that the sera would not recognize.
Considering the high similarity reported between Hsp83 sequences from T. cruzi and those from T. brucei and Hsp83 from L. m. amazonensis-being 95% and 85%, respectively (Rondinelli, 1994)-, along with the reported expression of L. infantum Hsp83 in cells transformed with pQE30 (Angel et al.; 1996), and thinking that the problem may be the residence time in the cell, a decision was made to clone into the latter vector directly, without waiting for the processes to verify the presence of the insert, that is, a “blind” strategy. Thus, the residence time in the bacterium would be reduced, which could prevent expulsion before the respective expression. In this case, it was possible to obtain positive results, with the appearance of a nearly 83 kDa protein in chromatography-eluted fractions from lysates of competent cells transformed E. coli M15.
Reviewing published articles on cloning genes encoding Hsp90 heat shock proteins in different parasitic models, either from genomic DNA or mRNA, reveals that the expression of proteins from the Hsp90 family of Trypanosomatids was possible through cloning in expression vectors. Examples of these vectors are the Hsp83 from L. braziliensis with the vector pET28a-TEV (Menezes-Souza et al.; 2014), Hsp90 from T. evansi with the vector pRSETA (Pallavi et al.; 2010), Hsp83 from Leishmania braziliensis from pET28a (Silva et al.; 2013), and Hsp83 from Leishmania infantum with pQE30 (Angel et al.; 1996). Among the expression vectors used in these studies, only pRSETA was more complex and employed in the current investigation. However, the results obtained were unsatisfactory, perhaps because the time lapses between subcloning in the expression vectors and expression were several weeks, and bacteria might have shed the recombinant plasmid due to its instability during that time. Using a direct “blind” subcloning strategy may have yielded better results in these vectors. Alternatively, this could be a particular characteristic of the Hsp83 of T. cruzi since it is strange that such an important molecule from a parasite causing serious public health problems has not had more cloning and expression studies, possibly due to the difficulty of its expression in prokaryotic systems.
In this sense, it is striking that during the review of the cloning and expression procedure of Plasmodium falciparum Hsp90, it was verified that the researchers directly cloned into the expression vector pET-23a and obtained the recombinant protein by using competent cells E. coli BL21 (Kumar et al.; 2003). Although the authors do not mention this aspect, they might have encountered circumstances similar to those discussed in this paper.
Regarding the expression of the recombinant protein, a low expression level and smaller bands were observed, which could be truncated forms of the protein, possibly due to bacterial attempts to degrade the recombinant protein. This degradation might occur due to sequence similarities, interfering with its metabolism (Huynh et al.; 1985). This could explain the limited observation of the recombinant protein.
The expected size of the recombinant protein according to the deduced amino acid sequence is 80.7 kDa + 1 kDa for the histidine tail; so, a molecular weight of nearly 82 kDa would be expected, which was observed in the results. It is important to note that HSP83 or HSP85 was so named since the native protein has a weight of 83-85 kDa. The small difference in size of nearly three kDa may be due to glycosylation of the native protein, as the prediction analysis of possible post-translational modifications of the sequence by Motif Scan of ISB-ISREC (Pagni et al.; 2001) evidenced four potential N-glycosylation sites. One of these sites can be considered functional since it is estimated that an N-glycosylation contributes an additional three kDa to the protein’s molecular weight (Obregon-Henao et al.; 2001).
As expected, problems were also encountered during the protein purification process because it was a large molecule of nearly 83 kDa, and its same weight was difficult to bind to the resin of the column only by a six histidine tail. It may be convenient to use another purification protocol or subclone the cDNA into a vector capable of binding to a larger fusion molecule that serves as a support and can then be eliminated, such as GST or MBP.
Considering the poor expression of hsp 83 and aiming to confirm that the band of nearly 83 kDa, whose expression increased with induction time, corresponded to the HSP83 protein of the parasite, a rabbit’s hyperimmune serum against T. cruzi epimastigotes (from the same isolate used in the present work) was used. This serum has been employed in several experimental protocols, with recognition of antigens with approximate molecular weights of 85, 80, 65, 62, 52, 38, 35, 34, 30, and 27 kDa in epimastigotes of the EP strain (Contreras et al.; 1998; Graterol et al.; 2013).
Immunoblotting analysis of the fractions obtained by nickel Ni-NTA column chromatography using hyperimmune anti-epimastigote serum facilitated the identification of a single band with an approximate molecular weight of 83 kDa, indicating, in this case, a reaction with the recombinant protein obtained in this study. Due to the low concentration of the recombinant protein used in the immunoblotting technique, the researchers decided to use a highly sensitive development system such as luminography, in particular with the Super Signal® West pico system, enabling the detection of immune complexes in pictograms. This explains the strong signal of the band observed on the X-ray film used to document the reaction.
Heat shock proteins have been shown to be immunogenic in various parasitic models (Polla, 1991), and most are important antigens recognized by patient sera. In addition, it has been reported that the immune response is directed to the less conserved areas of the protein, ensuring the specificity of the response (Skeiky et al.; 1995; Angel et al.; 1996). For example, specific anti-HSP90 antibodies have been documented in schistosomiasis (Johnson et al.; 1989), leishmaniasis (de Andrade et al.; 1992), and malaria (Zhang et al.; 2001). Immunodominant HSP70 antigens have also been reported in patients with hydatidosis (Colebrook & Lightowlers, 1997), schistosomiasis (Moser et al.; 1990), onchocerciasis (Rothstein et al.; 1989), leishmaniasis (de Andrade et al.; 1992; Wallace et al.; 1992; Skeiky et al.; 1995; Quijada et al.; 1996), Chagas disease (Engman et al.; 1989; Requena et al.; 1993) and malaria (Kumar et al.; 2003), and HSP60 in the case of leishmaniasis (Rey-Ladino et al.; 1997). Regarding low molecular weight HSPs, their diagnostic value in schistosomiasis and cysticercosis has been studied and confirmed (Nene et al.; 1986; Ferrer et al.; 2005).
It is important to note that no reactivity of the T. cruzi anti-epimastigote hyperimmune serum was observed with any other band of any size, only the 83 kDa, which would indicate that the possible truncated molecules lack the epitopes recognized by these antibodies. Using the complete protein to immunize rabbits and obtain hyperimmune sera that allow epitope mapping assays would be interesting.
Even at low concentrations, this recombinant protein HSP83 from T. cruzi can be used to elaborate a monospecific polyclonal antibody, which constitutes a very valuable tool to study the participation of this stress protein in the parasite’s differentiation processes, such as epimastigogenesis, metacyclogenesis, amastigogenesis, and trypomastigogenesis.
Conclusions
Despite the difficulties in the expression and purification of the recombinant protein, it could be expressed and identified with the hyperimmune serum. Therefore, this cloning and expression strategy is recommended for molecules with difficult cloning and expression.
Author contribution statement
All the authors declare that the final version of this paper was read and approved.
The total contribution percentage for the conceptualization, preparation, and correction of this paper was as follows: A.F.
33.3 %.; A.R.D.L. 33.3 % and E.F. 33.3 %.

