










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Costarricense de Cardiología
Print version ISSN 1409-4142
Rev. costarric. cardiol vol.3 n.2 San José Aug. 2001
Introducción
El nuevo milenio nos trae consigo un nuevo modelo o paradigma en la genética molecular cardiológica. Aunque se dice que la cardiología ha avanzado más en los últimos 50 años que en los 2000 años previos, es muy probable que avance aún más en las próximas dos a tres décadas. Este desarrollo vendrá de la genética molecular y de la aplicación de las técnicas de recombinación del ácido desoxirribonucleico. Ya han sido identificados todos los genes humanos; lo que ayudará a ligar miles de etiologías y factores de riesgo con sus respectivas enfermedades. Esto representa un nuevo modelo en la medicina, como se ilustra en el caso de la miocardiopatía hipertrófica (MH) familiar y las implicaciones que se deducen de ésta o también en el caso de los nuevos 50 genes ya identificados, responsables por enfermedades cardíacas. Actualmente se reconoce que la esperanza para la prevención y tratamiento de las enfermedades humanas no tiene precedente. Veinte enfermedades son las causantes del 80% de las muertes en el mundo occidental y son debidas a alteraciones en 100 a 200 genes, todos los cuales estarán disponibles en un par de años.
Fenotipo
La MH es una condición cardíaca primaria y usualmente familiar, caracterizada por hipertrofia ventricular con una fisiopatología compleja y gran heterogeneidad en su curso morfológico, funcional y clínico. Esta diversidad es enfatizada por el hecho de que la enfermedad se puede presentar en todas las fases de la vida, desde el recién nacido al viejo. El curso clínico es altamente variable, algunos pacientes permanecen asintomáticos durante toda la vida, otros desarrollan insuficiencia cardíaca severa y mueren prematuramente, y un grupo muere súbitamente a veces en ausencia de síntomas previos. La hipertrofia ventricular en esta condición es masiva, sin causa obvia, y se acompaña de alteración de la función diastólica y sistólica y tendencia a la muerte súbita. La distribución de la hipertrofia es generalmente en el ventrículo izquierdo (VI), principalmente en el tabique interventricular y en la pared ventricular izquierda, pero también se puede comprometer el ventrículo derecho (VD) y otras áreas menos típicas como la pared posterior u otras. La función sistólica puede ser hipercontráctil con disfunción diastólica, en muchos casos hay obstrucción del tracto de salida del ventrículo izquierdo (TSVI) y en algunos del tracto de salida del VD.
La hipertrofia diagnósticada por ecocardiograma es lo que convencionalmente se conoce como la expresión fenotípica de la MH y es lo que se usa para ligar a locus genéticos.
Prevalencia y mortalidad.
Observaciones recientes sugieren que la prevalencia de MH en la población general es más alta (alrededor de 1 en 500) que lo que se había pensado. La condición por lo tanto impresiona ser una malformación genética común del corazón. Como la historia natural de la enfermedad es muy variable se hace difícil conocer su espectro total, sobre todo por el hecho de que los pacientes que son referidos a centros de atención terciaria son preferencialmente aquellos más enfermos. De manera que la mortalidad anual reportada por los hospitales (3 a 4 % en general y hasta un 6% en niños) es sustancialmente más alta que la reportada en poblaciones no seleccionadas (1 % o menos). Estos sugiere que en una proporción sustancial de pacientes, la enfermedad tiene un curso clínico más favorable de lo que se pensó antes. En MH la muerte, particularmente cuando es súbita se da sobre todo en personas jóvenes. En un estudio reciente, en una gran serie de 744 pacientes de un grupo internacional, con toda clase de población portadora de esta enfermedad, fallecieron 86 pacientes (12%) en un período de 8+/-7 años. Se presentaron tres distintos modos de muerte: (1) súbita e inesperada (51%; edad 45+/-20 años); (2) insuficiencia cardíaca progresiva (36%; edad, 56+/-19 años); y (3) accidente vascular cerebral asociado con fibrilación atrial (13%; edad, 73+/-14 años). La muerte asociada a accidente vascular cerebral ocurrió desproporcionadamente en pacientes mayores. Como dato interesante y que va a ser una constante en muchos estudios, de los 45 pacientes que murieron súbitamente, la mayoría (71%) no tenían síntomas. Aunque la mayoría de las muertes súbitas ocurrieron en adolescentes y en adultos jóvenes, también se presentaron más tarde en la vida.
Comportamiento clínico.
La mayoría de los pacientes con MH están asintomáticos o solo ligeramente sintomáticos y frecuentemente se identifican durante el estudio de algún familiar con la enfermedad. Desafortunadamente la primera manifestación clínica en los individuos asintomáticos puede ser la muerte súbita. En cuanto a los pacientes con esta enfermedad que tienen síntomas, en general se pueden agrupar en cuatro grupos de acuerdo a cual manifestación clínica predomine en ellos. El grupo en que predomina la disfunción diastólica, el que predomina la isquemia, la obstrucción intraventricular y en los que predominan las arritmias y el riesgo de muerte súbita, y lógicamente mezclas de estos. Los síntomas fundamentales son disnea de esfuerzo, angina y síncope. Los hallazgos del examen físico más característicos son el pulso carotídeo bisferiens y el soplo sistólico paraesternal izquierdo. El pulso bisferiens es una característica frecuente tanto en la MH de tipo obstructivo como en la no obstructiva. El soplo es rasposo holosistólico paraesternal izquierdo, aumenta durante la maniobra de Valsalva, y sugiere MH obstructiva. Como la forma no obstructiva de MH es más frecuente, la característica bien descrita de obstrucción dinámica del tracto de salida, tal como un soplo sistólico fuerte no se requiere para el diagnóstico. También se presenta un soplo de eyección apical con un pico de intensidad sistólico tardío como un signo fonocardiográfico indicativo de obstruction dinámica, medio ventricular izquierda. Este soplo debe ser diferenciado del de regurgitación mitral en aquellos pacientes que lo tienen. La presencia de un cuarto ruido (S4) es un hallazgo común en MH. La MH es una enfermedad cardíaca primaria típica caracterizada por función diastólica anormal, debido tanto a relajación prolongada como compliance disminuida, con contribución aumentada de la sístole atrial al llenado ventricular.
Pacientes con miocardiopatía no obstructiva
En estos casos la función diastólica del (VI) es el principal determinante de los síntomas. Hay disminución de la capacidad de ejercicio dado por alteración en la función pasiva diastólica del VI. Esto se ha estudiado con Ecocardiografía modo M del atrio izquierdo y por las velocidades de flujo venoso pulmonar medido por Doppler. La disnea es el síntoma más común (90% de los pacientes sintomáticos). La disfunción diastólica de los ventrículos puede ser mejorada prolongando el período de llenado diastólico, acortando el período de relajación isovolumétrica con drogas beta bloqueadoras y con bloqueadores de calcio, o tal vez alterando el período de activación atrioventricular con marcapaso DDD. Angina es el otro síntoma más común y ocurre en algún momento en el 75% de los pacientes. Hay una variedad de mecanismos que llevan a angina. Por lo menos en parte la angina pectoris se presenta como parte de un desbalance entre la oferta de oxígeno para un músculo con una demanda aumentada por la gran cantidad de masa miocárdica. También hay anormalidades de las pequeñas arterias coronarias y mucha fibrosis intramiocárdica y perivacular. También se presenta fatiga y palpitaciones dados por la variedad de arritmias que acompañan a la enfermedad. El ejercicio puede empeorar muchas de estas manifestaciones. Se ha relacionado también la aparición de síncope en aquellos pacientes que tienen cavidad ventricular pequeña.
Forma apical
La MH apical es caracterizada por hipertrofia primaria localizada exclusivamente en el ápex del VI. El diagnóstico se hace con una combinación única entre los hallazgos ecocardiográficos y los del electrocardiograma (ECG), que consiste en ondas T gigantes invertidas y alto voltaje de la onda R en las derivaciones precordiales. La característica fisiológica mayor de la MH apical es disfunción diastólica, debido a rígidez anormal del VI durante la diástole, lo que resulta en dificultad al llenado ventricular. En estos pacientes, al igual que en todos aquellos con MH la principal patología es relajación inadecuada de los ventrículos; lo que se puede presentar como insuficiencia cardíaca congestiva. Por el trastorno de llenado, un régimen de diuréticos y agentes inotrópicos puede empeorar el curso clínico.
Forma obstructiva
En este caso la obstrucción en el TSVI es el determinante más importante de los síntomas clínicos. Esto produce la generación de un gradiente de presión intraventricular, que característicamente es dinámico o sea que aumenta de acuerdo al aumento del trabajo del corazón. Los síntomas más frecuentes son la angina de esfuerzo y el síncope de esfuerzo. El gradiente de presión se mide por Doppler en el sitio de la obstrucción.
Diagnóstico:
Electrocardiograma.
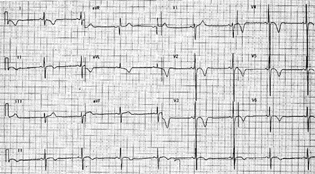
En la mayoría de los pacientes con MH el electrocardiograma es anormal. Las alteraciones más comunmente observadas son: hipertrofia ventricular izquierda, ondas Q patológicas, ondas T negativas, y ondas T negativas gigantes, y elevación del segmento ST. La figura 4 muestra un ECG de un paciente con MH de la consulta externa del Hospital México. Ocasionalmente, la localización de la hipertrofia ventricular puede ser sugerida por el electrocardiograma. El monitoreo de Holter se debe hacer siempre en estos pacientes ya que nos permite evaluar uno de los factores pronósticos más importantes que es la taquicardia ventricular no sostenida. Es común la depresión del segmento ST en pacientes con MH y arterias coronarias permeables. La inducción de depresión del segmento ST con ejercicio que se ve en la MH no obstructiva se asocia con disfunción sistólica en esfuerzo. Se ha encontrado que el intervalo QT y el QTc así como la dispersión de QT (definido como la diferencia entre el QT máximo y el mínimo en las doce derivaciones) es mayor en pacientes con MH comparado con subjectos normales. Los pacientes con MH con frecuencia muestran un QTc prolongado (> 440 ms). Además, el grado de hipertrofia ventricular izquierda correlaciona con el QTc máximo. La presencia de un QT prolongado con aumento de la dispersión regional puede estar asociado con la aparición de arritmias ventriculares serias y muerte súbita en MH. También se ha asociado la MH con el síndrome de Wolff-Parkinson-White (WPW) y ya se ha mapeado una alteración en el cromosoma 7 de los pacientes que tienen MH concomitantemente con WPW.
Estudio electrofisiológico:
El rol de los estudios invasivos electrofisiológicos es controversial ya que las anormalidades producidas por estimulación programada tienen muy baja especificidad.
Ecocardiografía:
El diagnóstico clínico de MH ha estado basado en la identificación por ecocardiografía bidimensional de la condición morfológica más característica de la enfermedad que es engrosamiento de la pared ventricular izquierda (usualmente asimétrica en distribución), asociada con una cámara no dilatada, en ausencia de otra enfermedad cardíaca o sistémica capaz de producir esa magnitud evidente de hipertrofia (hipertensión sistémica o estenosis aórtica). Los pacientes con MH muestran un amplio espectro de grosores de la pared del ventrículo izquierdo. La magnitud del grosor de la pared miocárdica encontrado en los pacientes que se identifican clínicamente (promedio de 20 a 22mm y hasta 60mm) generalmente permite el diagnóstico inequívoco, aunque grados más modestos de hipertrofia (15 a 20mm) se encuentra con frecuencia, particularmente durante el mapeo de pedigree y también en los ancianos. El gran problema es en los casos de expresión fenótipica más sútil con grosor entre (13 a 15mm) sin obstrucción al TSVI, cuando se crea ambigüedad diagnóstica y con frecuencia hay dilemas clínicos. Cuando esto ocurre en atletas altamente entrenados, la diferenciación de hipertrofia benigna puede ser difícil pero se puede resolver con una evaluación no invasiva clínica o con mapeo genético donde es posible. Por último no todos los individuos que tienen la genética tienen hipertrofia en toda la vida. La hipertrofia a veces no aparece hasta la adolescencia y a veces hasta que se haya completado toda la maduración. Aunque parece que el proceso de remodelado está usualmente completo a los 18 años, unos pocos adultos afectados genéticamente con penetrancia variable (y particularmente en los defectos genéticos en los que hay mutación en los genes para la proteina C que liga la miosina) pueden mostrar muy poca o ningún grado de hipertrofia (menos de 13mm de grosor). Consecuentemente es posible que el proceso hipertrófico se pueda retrasar hasta más allá de la mitad de la vida o más. Hay características ecocardiográficas como el movimiento anterior sitólico de la vávula mitral, o el cierre parcial prematuro de la vávula aórtica que se observan por ecobidimensional y modo M pero solamente en los casos cuando hay obstrucción al TSVI. A un grupo de 60 casos con MH se les efectúo ecotransesofágico y se estudiaron las características del aparato valvular mitral. Se encontró que las valvas son más largas, tanto en las MH obstructivas como en las no obstructivas. También se observó contacto mitral septal en alrededor del 85% de los pacientes con obstrución y hasta en un 10% de los que no tienen obstrucción. Las estructuras que participaron en el fenómeno de movimiento anterior sistólico de la válvula mitral fueron: la porción distal de la valva anterior de la mitral (77.5%), ambas valvas mitrales (18.4%) y cuerdas anómalas (4.1%). En 5 pacientes la obstrucción aún más allá de las cuerdas. Se observó regurgitación mitral en 43 pacientes; en 37 de ellos el jet se dirigía posteriormente en sístole tardía. Los pacientes MH tienen válvulas mitrales más largas y con frecuencia presentan anormalidades asociadas sugiriendo que esta enfermedad no se confina al miocardio y que el largo de las valvas no es el único determinante de obstrucción. En casi el 80% de los pacientes el movimiento anterior sistólico fue producido por la parte distal de la valva anterior, lo que resultó en coaptación incompleta en mediosístole. La secuencia de eventos sistólicos fue eyección/obstrucción/regurgitación. También es importante el análisis del perfil del flujo venoso pulmonar y del influjo mitral. En un estudio de 80 pacientes con MH a los que se les estudió el flujo venoso pulmonar por ecocardiograma transoesofágico; éstos pacientes muestran aumento del flujo reverso pulmonar y tiempo de desaceleración de la onda diastólica prolongado. Sin embargo se dió una gran variación en los patrones del flujo venoso, 30 pacientes (37.5%) tenían flujo mitral pseudonormal por lo que se considera importante además de valorar el tamaño del atrio izquierdo también valorar el flujo pulmonar sobre todo en aquellos que tienen flujo mitral pseudonormal. La relación entre los tiempos de tránsito de la onda A y la onda E del influjo mitral, dentro del VI (T(e)/T(a) se relaciona cercanamente con la relajación del VI, su relajación en diástole tardía, y las presiones de llenado, dando mucha información sobre la función diástolica. En MH, se pueden determinar en forma confiable las presiones de llenado del VI, midiendo la relación entre la velocidad de la onda E y la velocidad de propagación de la onda E a nivel anular, medido por modo M color (E/Ea).
Diagnóstico diferencial
Algunas veces es difícil diferenciar la MH de la cardiopatía hipertensiva. Algunos pacientes con MH muestran hipertrofia simétrica, mientras pacientes con cardiopatía hipertensiva algunas veces tienen hipertrofia septal asimétrica. Se ha usado un ácido graso de cadena larga marcado con radioiodo para visualizar el metabolismo alterado de los ácidos grasos en el miocardio. La carnitina es la sustancia esencial para la beta oxidación de los ácidos grasos de cadena larga. Los niveles de carnitina libre en el suero están elevados en MH y correlacionan con la severidad del metabolismo de los ácidos grasos. Los niveles de carnitina pueden ayudar a diferenciar estas dos enfermedades. El entrenamiento regular intensivo puede causar aumentos leves en el grosor de la pared del VI. Aunque el grado de hipertrofia del VI es típicamente menos de la que se ve en MH, los estudios genéticos han mostrado que una substancial minoría de pacientes con MH tienen un grosor de pared en el mismo rango. La diferenciación entre hipertrofia fisiológica y patológica en esta "zona gris" puede ser problemática usando ecocardiografía. La prueba de esfuerzo con análisis metabólico facilita la diferenciación entre hipertrofia fisiológica y la MH. Un pVO2 >50 ml/kg/min o >20% sobre el máximo predicho diferencia el corazón de atleta. Los pacientes con MH exhiben VO2 máx, significativamente más bajos así como el umbral anaeróbico y la oximetría de pulso comparado con controles. El reconocimiento de EAC coexistente en pacientes MH puede ser muy difícil utilizando métodos no invasivos que se basan en cambios electrocardiográficos o en defectos de perfusión. En un estudio con 88 pacientes con MH; se llevó a cabo ecocardiografía de estrés con altas dosis de dipiridamol (hasta 0.84 mg/kg en 10 minutos). A 60 pacientes se les hizo arteriografía coronaria independientemente de los resultados del test. Asumiendo que el único criterio de positividad fuera disquinesia transitoria, y definiendo EAC como > o = 50% estrechez del diámetro en por lo menos un vaso coronario mayor, se encontró una sensibilidad del 71%, una especificidad del 100%, un valor predictivo positivo 100%, y una confiabilidad diagnóstica para EAC determinada por arteriografía coronaria del 93%. Se concluye en este estudio que este método es confiable para el diagnóstico no invasivo de EAC concomitante con MH. Sin embargo en otro estudio en el que se hizo ecocardiografía de estrés con dobutamina o con ejercicio en pacientes con MH; se encontró disfunción sistólica en el 50% de los pacientes con MH y sin EAC. En estos pacientes, se encontraron anormalidades de la contractilidad regional en los segmentos hipertrofiados.
Genotipo:
Hasta el año 2000, se conocen alteraciones en nueve genes que codifican para las diferentes proteínas del sarcómero. La contracción cardíaca es producida por la interacción cíclica del "motor molecular"; o sea de la proteína miosina con el filamento actina. Esto consume ATP como la fuente de energía que produce tensión o acortamiento. La miosina de cadena pesada contiene los sitios donde se liga la actina y el ATP y representa el motor molecular de la contracción muscular. En el corazón humano se expresan dos genes diferentes para la miosina de cadena pesada (alfa y beta) con características bioquímicas distintas. La miosina alfa de cadena pesada confiere mayor actividad ATPasa y velocidad de acortamiento más alta que la beta-miosina de cadena pesada. La función motora es regulada por las isoformas de miosina de cadena liviana. En MH se han encontrado mutaciones en todas las subunidades de miosina. También mutaciones en los genes que codifican para la troponina cardíaca T pueden causar MH familiar, la troponina T es la subunidad que permite que se lige la tropomiosina, lo cual se requiere para que el filamento delgado regule la contracción. Esta se ocupa de un rol único en el sistema de proteínas reguladoras porque interactúa con la troponina C y con la troponina I del complejo troponina y las proteínas del filamento delgado, tropomiosina y actina. La troponina T se usa clínicamente por su potencial para detectar daño miocárdico. Las mutaciones de la troponina T son muy importantes porque se han encontrado en familias donde han habido muertes súbitas, en ausencia de aumento en la masa ventricular a nivel clínico o macroscópico.
Se han encontrado mutaciones en nueve genes; pero sin embargo solamente en la mitad de los pacientes con MH, lo que sugiere que todavía hay otros que no se han descubierto .
Estos son las nueve mutaciones que se conocen:
1- beta miosina de cadena pesada.De los nueve genes alterados conocidos, hay cuatro más comunes que tienen daños característicos: 1) el de la miosina mutante , 2) el de la troponina T , 3) la alfa tropomiosina, y 4) el gen para la proteína C que liga la miosina.
2- alfa-tropomiosina.
3- troponina cardíaca T.
4- troponina I.
5- La proteina C que liga la miosina.
6- Los dos genes de miosina regulatoria de cadena liviana.
7- La miosina esencial de cadena liviana.
8- La actina cardíaca.
9- La titina.
Además, se han reportado mutaciones en los dos genes que codifican las cadenas de miosina livianas en lo que parece ser una forma rara de MH. Esta complejidad etiológica es todavía más complicada por lo que se conoce como heterogeneidad intragénica, o sea que se han identificado más de 50 mutaciones causantes de enfermedad en estos genes del sarcómero. Por lo tanto el defecto molecular preciso responsable por MH con mucha frecuencia varía en pacientes no relacionados. Como se ve ésta es una enfermedad heterogénea genéticamente y así va a ser también desde el punto de vista clínico. El conocimiento de mutaciones subyacentes ha clarificado la interpretación de las características clínicas, y ha mostrado que casi todos los casos son familiares y que se requieren criterios diagnósticos menos exigentes entre las familias. El diagnóstico y el pronóstico se han refinado. Esta heterogeneidad de la enfermedad se acentúa por el hecho de que afecta a pacientes de todas las edades.
Patología
Desde la descripción anatómica moderna de Teare en 1958 , la hipertrofia ventricular izquierda se ha considerado tradicionalmente como el marcador anatómico macróscopico y el determinante probable de muchos de sus características clínicas y consecuencias en este tipo de pacientes. Debido a que la cavidad ventricular izquierda es usualmente pequeña o normal, el aumento en masa ventricular es debido casi exclusivamente a aumento de la pared ventricular. Al exámen histológico lo más común es desorganización de las miofibrillas. Además, se ha demostrado en corazones de autopsias que las arterias intramiocárdicas son displásticas. Tanto el grosor como la fibroelastosis de la media es casi el doble de lo normal. La túnica íntima también se encuentra engrosada frecuentemente y el lumen relativamente reducido. EL músculo ventricular se engruesa con un cuadro histológico de hipertrofia irregular, con un acomodo desorganizado de miocitos. Hay formación y alineamiento anormal de miofibrillas lo que les da una forma anormal a las células. Mientra todos los miocitos llevan el gen, la selectividad regional de la hipertrofia no se puede explicar. La expresión fenotípica de la enfermedad por ejemplo en los miembros afectados de una misma familia, todos los cuales son heterocigotos para la misma anormalidad genética es muy variada. El aumento en el peso total del corazón va desde casi normal hasta a más de 800 gramos. En un estudio de 75 casos el compromiso ventricular fue difuso y simétrico en el 42%. La forma asimétrica más común comprometió la región anteroseptal (31%) pero casos esporádicos comprometieron solamente la pared posterior o lateral. Una minoría de casos (9.5%) no mostró engrosamiento macroscópico de la pared. Se encontró fibrosis generalmente asociada con cambios displásticos en la media de las pequeñas arterias intramiocárdicas lo que puede llevar a una pared ventricular que simula una miocardiopatía dilatada. En una tercera parte de los casos, se encontró un parche subaórtico de engrosamiento endocárdico en el septum ventricular, debido a contacto con la valva anterior de la mitral. El mecanismo subyacente en paro cardíaco en MH es intrigante. Desde el punto de vista clínico se ha incriminado la isquemia miocárdica por mucho tiempo, particularmente en el joven. En un estudio de 274 muertes súbitas cardiovasculares en el joven (< o = 35 años), 19 (7.0%), 14 hombres y 5 mujeres, edad media de 23 años, tenían MH. Se encontraron en estos 19 pacientes, hallazgos patológicos de daño isquémico, ya sea agudo o subagudo o en forma de cicatrices fibróticas. Los autores encuentran evidencia de que ocurre isquemia en la historia natural de MH lo que puede contribuir a que se desarrolle inestabilidad eléctrica y arritmias fatales. En cuanto a la investigación de muerte súbita cada vez más los patólogos se enfrentan con una complejidad mayor en cuanto a las enfermedades que causan muerte súbita natural en ausencia de EAC. Una proporción significativa de tales muertes naturales son debidas a enfermedades familiares del músculo cardíaco como MH y displasia arritmogénica del VD. Las características fenotípicas de ambas son más amplias de lo que se había pensado y macroscópicamente los corazones pueden parecer normales. Solo la histología detallada puede identificar tales casos. En Inglaterra, ocurren alrededor de 200 muertes súbitas al año en jóvenes aparentemente bien acondicionados, en los que los exámenes de toxicología y el estudio detallado estructural de corazón es negativo. Estos casos se explican por defectos genéticos de los canales iónicos (intervalo QT largo). Cuando se investiga un caso de muerte súbita sin causa aparente el estudio de los familiares podría ayudar.
Muerte súbita.
Uno de los problemas más difíciles para el médico y el paciente es el no poder predecir y/o evitar eventos adversos, como en el caso de la muerte súbita. Como se sabe, la presencia o ausencia de un gradiente en el TSVI, es un pobre predictor de alto riesgo de muerte súbita. Muchos estudios han analizado como identificar pacientes en alto riesgo. Los factores que mejor identifican alto riesgo de muerte súbita son: historia previa de muerte súbita o síncope, inducción en adultos de taquicardia ventricular sostenida, la presencia de taquicardia ventricular no sostenida en pacientes asintomáticos, isquemia asociada con hipotensión en niños, y una pobre fracción de eyección. En cuanto a la extensión de hipertrofia cardíaca se ha dicho que también es un pobre predictor excepto cuando es masiva. Sin embargo, en un estudio muy reciente en 480 patientes consecutivos con MH, se evaluó la relación entre la magnitud de hipertrofia ventricular izquierda y mortalidad. Los pacientes se categorizaron en 5 subgrupos de acuerdo al grosor máximo de pared: 15 mm o menos, 16 a 19 mm, 20 a 24 mm, 25 a 29 mm, y 30 mm o más. Las edades oscilaron entre 1 a 89 años (media 47). Se les dió seguimiento por un período de 6.5 años en promedio, 65 de los 480 pacientes (14 %) murieron: 23 súbitamente, 15 de insuficiencia cardíaca, y 27 de causas no cardíacas o accidente vacular cerebral. El riesgo de muerte súbita aumentó progresivamente y en relación directa con el grosor de la pared (P=0.001), en un rango de 0 por 1000 personas-año (95 % intervalo de confidencia, 0 a 14.4) para grosor de 15 mm o menos a 18.2 por 1000 persona-año (95 % intervalo de confidencia, 7.3 to 37.6) para paredes mayores de 30 mm o más y casi dobla entre cada grupo con el que sigue. El riesgo acumulado en 20 años después de la evaluación inicial era cerca de 0 para pacientes con grosores de 19mm o menos pero casi 40 % para grosores de 30mm o más. Al comparar los pacientes con hipertrofia extrema con otros subgrupos, estos fueron los más jóvenes (edad promedio, 31 años), y la mayoría (41 de 43) tenían síntomas leves o no tenía síntomas; de los 12 pacientes que tenían menos de 18 años en la evaluación inicial, 5 murieron súbitamente. En MH, la magnitud de la hipertrofia está directamente relacionada con el riesgo de muerte súbita y es un predictor fuerte e independiente de pronóstico. Los pacientes jóvenes con hipertrofia extrema, aún aquellos con pocos síntomas o sin ellos, parecen tener un riesgo substancial a largo plazo y merecen la consideración de intervenciones que prevengan la muerte súbita. La mayoría de los pacientes con hipertrofia leve están en bajo riesgo. Estos otros elementos mencionados si tienen valor pronóstico; la historia familiar de muerte súbita, el síncope recurrente, la taquicardia ventricular no sostenida, y una respuesta anormal de la presión sanguínea. Esta última sobre todo tiene valor predictivo negativo. La mayoría de los pacientes no tienen estas características y pueden por lo tanto se categorizadas como de bajo riesgo para muerte súbita. Los pacientes que requieren anestesia general y que no se conocen portadores de la enfermedad pueden presentar paro cardíaco intratable. Por ejemplo una leve hipotensión causada por anestesia epidural puede tener un efecto devastante. Por esto es importante hacer el diagnóstico temprano y dar observación cuidadosa durante el período perioperatorio.
Profilaxis contra la muerte súbita:
Todavía no se han sugerido terapias nuevas por los hallazgos de genética molecular. La diversidad de anormalidades en las proteínas contráctiles todavía obscurece el camino hacia la enfermedad. Un componente del proceso parece ser hipertrofia compensadora que es inicialmente, una respuesta adaptativa a contractilidad disminuida (ej: el caso de miosina mutante). Otras mutaciones, sobre todo aquellas que afectan la tropo-nina T cardíaca, tienen poco efecto en generación de fuerza y por lo tanto causan menos hipertrofia pero resultan en uso ineficiente de energía. Estas mutaciones son particularmente llamativas, porque se asocian con hipertrofia miocárdica sútil pero con un riesgo particularmente alto de muerte súbita. Estos hallazgos sugieren que hay diferentes vías que llevan a hipertrofia y a muerte súbita potencial e ilustran el poder potencial del diagnóstico molecular directo; ya que una proporción de personas llevan mutaciones pero no tienen evidencia de MH por criterios convencionales, y como se ha mencionado sin embargo; dependiendo del tipo de mutación, algunas de estas personas estan aún en riesgo de muerte súbita. Un problema por definir es el del diagnóstico preclínico en familias ya sea por clínica o por estudio del DNA. Aunque esto puede estar indicado para pronóstico, tal abordaje sería apropiado solamente en el caso de que el tratamiento mejorara la sobrevida, o si se pudiera evitar la complicación más catastrófica que es la muerte súbita. Aunque se ha asumido que son varios los mecanismos que conllevan a los pacientes a muerte súbita: fibrilación atrial, bradicardia, isquemia miocárdica, vasodilatación periférica durante ejercicio y arritmias ventriculares y supraventriculares que llevan al colapso cardíaco; hoy se sabe que una arritmia ventricular espontánea es la culpable. Sin embargo, debido a la heterogeneidad genética de este desorden, diferentes grupos de pacientes pueden tener diferentes riesgos de muerte súbita. Estudios de cohorte que han comparado pacientes con MH quienes han sido tratados con amiodarona a bajas dosis vs controles no tratados, sugieren que el tratamiento a largo plazo es parcialmente protector. Tratamiento con altas dosis de beta-bloqueadores puede conferir proteción. Puesto que hay un exceso de muerte súbita durante o inmediatamente después de hacer ejercicio, la mayoría de los médicos recomiendan a los pacientes con MH, el evitar deportes competitivos o ejercicio intenso. Sin embargo no hay datos que indiquen que los pacientes que no lo hacen tienen un riesgo reducido.
Manejo de la muerte súbita en MH.
Los datos al respecto se basaron inicialmente en estudios retrospectivos de prevención secundaria en pacientes que sobrevivieron paro cardíaco o presentaron taquicardia ventricular sostenida. Entre los sobrevivientes de fibrilación o taquicardia ventricular sostenida que causa síntomas severos el desfibrilador implantable es superior a las drogas antiarrítmicas de la clase 3 (amiodarona principalmente) para aumentar la sobrevida. El estudio AVID (The antiarrhythmics versus implantable defribrilators in patients resuscitated from near fatal ventricular arrhyhmias) publicado en 1997, se realizó en 56 centros. Se estudiaron 1013 pacientes resucitados de fibrilación ventricular casi fatal o a quienes se les había efectuado cardioversión eléctrica por taquicardia ventricular con compromiso hemodinámico. Un grupo se trató con implantación del cardiovertor-desfribrilador eléctrico y el otro recibió una droga clase 3 (Amiodarona) por un período de casi 4 años. De los 1013 pacientes (el 45% de los cuales habían tenido fibrilación ventricular, y el 55 % taquicardia ventricular) 507 fueron asignados al azar a tratamiento con el desfibrilador cardiovertor implantable y 509 a drogas antiarrítmicas. El objetivo primario del estudio era evaluar la mortalidad total. El seguimiento fue completo en el 99.7 % de los casos y la sobrevida mejor en los pacientes con desfibrilador implantable que en el grupo bajo amiodarona, 89.3% vs 82.3% en el primer año, 81.6 vs 74.7 a los dos años, y 75.4% vs 64.1% a los tres años. El estudio concluye que el desfibrilador eléctrico debe usarse como tratamiento de primera línea en los pacientes resucitados de estas arritmias. Este estudio muestra que los pacientes con MH que sobreviven un episodio de taquicardia ventricular o de fibrilación ventricular permanecen en alto riesgo de eventos recurrentes. La terapia con el cardiovertor-desfibrilador implantable parece ofrecer el mejor beneficio potencial con respecto a sobrevida en esta temible condición.
Los tratamientos que limitan el trabajo cardíaco, los que resultan en una mejor utilización de la energía o que aumenten su aporte pueden modificar la enfermedad. Mientras tanto el tratamiento va dirigido a prevenir complicaciones. En el año 2000 Maron et al, presentó los resultados del estudio retrospectivo más grande, multicéntrico, que provee soporte definitivo para el uso del desfibrilador implantable para prevención secundaria y también para prevención primaria en pacientes seleccionados de alto riesgo. Este estudio trae buenas noticias pues pudo documentar los eventos fatales y lo que habría pasado sí no se hubiera descargado el desfibrilador. Se probó la eficacia del cardiovertor-desfibrilador implantable para prevenir muerte súbita en 128 pacientes con MH a quienes se les juzgó como de alto riesgo de sufrirla. Al momento de la implantación del desfibrilador, los pacientes tenían de 8 a 82 años de edad (promedio [±DS], 40±16), y 69 pacientes (54 %) tenían menos de 41 años. El promedio de seguimiento, fue un período de 3.1 años. Los desfibriladores fueron activados apropiadamente en 29 pacientes (23 %), se descargaron para proveer choque de desfibrilación o para cardiovertir una taquicardia, con restauración del ritmo sinusal; la edad promedio a la que ocurrió la intervención fue de 41 años. El porcentaje de descargas apropiadas fue el 7% por año. Un total de 32 pacientes (25%) tuvieron episodios de descargas inapropiadas. En el grupo de 43 pacientes que recibieron desfibriladores para prevención secundaria (después de paro cardíaco o taquicardia ventrícular sostenida), los aparatos fueron activados apropiadamente en 19 pacientes (11 % por año). De los 85 pacientes a quienes se les puso el implante en forma profiláctica por los factores de riesgo (para prevención primaria), 10 tuvieron intervenciones apropiadas (5% por año). El intervalo entre la implantación y la primera descarga apropiada fue altamente variable, pero en 6 pacientes fue muy prolongada (4 a 9 años). En todos los 21 pacientes con datos electrocardiográficos registrados y con descargas apropiadas, las intervenciones fueron provocadas por taquicardia o fibrilación ventricular. En este estudio, a una tercera parte de los pacientes se les implantó el aparato para prevención secundaria. En este grupo, el porcentaje anual de descargas apropiadas fue el 11 %, con radio acumulativo del 75% a 10 años. Una cantidad de intervenciones impresionantemente más alta ocurrió en los primeros 4 meses después de la implantación, lo que confirma que los pacientes con miocardiopatía tienen períodos inestables. Pero sin embargo también se dió mucha recurrencia y eventos tardíos. Estos resultados, al igual que la edad promedio relativamente joven de los pacientes, sugiere que el grado de descargas apropiadas puede ser más alto cuando el desfibrilador se usa para prevención secundaria, en pacientes con MH que cuando se usan en pacientes con taquicardia inducible después de un infarto agudo del miocardio. Ahora todavía de más importancia son los resultados en los dos tercios de pacientes que recibieron el desfibrilador para prevención primaria. Es importante notar que aquí se incluyeron pacientes para la implantación con criterios no muy estrictos como por ejemplo, la típica persona encontrada durante un chequeo por un familiar con antecedente de muerte súbita. Los factores de riesgo principales estaban presentes cada uno en aproximadamente un tercio a la mitad de los 85 pacientes: síncope, historia familiar de una o más muertes súbitas, y taquicardia ventricular no sostenida. Algunos de los pacientes quienes recibieron descargas que les salvaron la vida se habían metido en el estudio solamente por taquicardia ventricular asintomática no sostenida detectada por monitoreo de Holter. En este grupo de prevención primaria, el porcentaje anual de descargas apropiadas fue del 5%. El porcentaje acumulativo de descargas alcanzó una meseta en aproximadamente 20%. Se extrapola que a 10 años será del 50%. Si ésto se logra probar con estudios de más duración entonces no quedará duda, de que los costos y los riesgos de usar un desfibrilador implantable serán muy superados por el número de vidas salvadas. Ahora como el desfibrilador termina los eventos fatales y no los está previniendo, con este estudio se documentó la muerte súbita en forma muy efectiva, en pacientes con MH. En todos los 21 pacientes con desfribriladores que se descargaron en forma apropiada y que registraron y almacenaron datos electrocardiográficos, intracardíacos el evento desencadenante fue taquicardía o fibrilación ventricular. Aún se debate si fué por isquemia regional, por compromiso del metabolismo energético cardíaco, por conducción anormal debido a desorden miofibrilar, o a alguna combinación de estos factores que se inicia o se exacerban tales eventos, ahora sí conocemos el mecanismo de la mayoría de las muertes súbitas. En más del 70 % de los pacientes con descargas apropiadas, hubo múltiples descargas y en el 21 % de los pacientes con descargas apropiadas, la primera descarga ocurrió 4 o más años después de la implantación. La mitad de las intervenciones apropiadas ocurrieron a pesar de terapia antiarrítmica por lo que la protección dada por el defibrilador implantable fue considerablemente superior.
Todavía quedan mucha cosas por resolver por lo menos en lo que a prevención primaria se refiere por que lo difícil es la identificación precisa de los pacientes en riesgo de muerte súbita. Se ha sugerido que se debe crear un registro internacional para documentar las descargas después de implantación para cada uno de los factores de riesgo. Idealmente, debería incluirse información molecular puesto que la mutación subyacente será predictiva; es más, la MH atribuible a los diferentes genes alterados es muy probable que estén asociados con diferentes factores de riesgo fenotípicos. Así se podrá tomar ventaja también de los avances en ciencias básicas.
Muerte súbita en atletas y deportistas
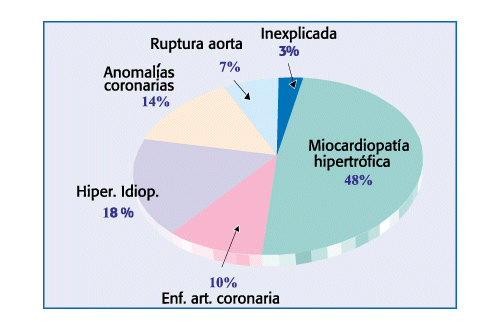
La MH es la causa más común de muerte súbita en individuos que parecen estar sanos. El riesgo es más alto entre los 14 y los 35 años, precisamente la edad de la mayoría de los atletas. La figura 5 muestra las causas de muerte súbita en los atletas menores de 35 años. En 80 pacientes que murieron de muerte súbita durante e inmediatamente después del ejercicio; se identificaron las lesiones que causaron la muerte y se encontró que son las mismas que se encuentran en atletas profesionales. Llama la atención que a pesar de que esta enfermedad es la causa más frecuente de muerte súbita en atletas; aún no se consideren necesario ni el ECG ni la prueba de esfuerzo, para evaluar los atletas previo a participación; en aquellos pacientes que tienen historia y exámen físico negativo. Sobre todo tomando en cuenta que las condiciones específicas que excluirían o limitarían la participación atlética incluyen la MH, y el síndrome de intervalo QT largo; y que los criterios diagnósticos convencionales aunque son muy específicos no son muy sensitivos para la detección clínica de la enfermedad. En Italia se les dio seguimiento a más de 35.000 jóvenes por más de 20 años, a quienes se les hizo un exámen preparticipación en deportes. Ellos encontraron que la MH fue una causa poco común de muerte en los atletas jóvenes competitivos y sugieren que fue la identificación y la descalificación de los atletas afectados lo que probablemente previno la muerte súbita.
Tratamiento
El tratamiento más efectivo de los pacientes sintomáticos con MH todavía está en discusión. A la hora de tomar la decisión de manejo de estos pacientes, es importante reconocer la heterogeneidad de esta enfermedad y dirigir la terapia a las anormalidades predominantes. Las características genéticas y clínicas tan diversas de la MH hacen imposible definir lineamientos precisos de manejo. Por otro lado hay que tomar en cuenta, que una cosa es el tratamiento de los síntomas para mejorar calidad de vida y otra y quizá la más importante es la identificación de los pacientes que están en alto riesgo de muerte súbita y requieren terapia agresiva. Ambas deben abordarse con estrategias diferentes.
Opciones de tratamiento incluyen:
A) Terapia médica.
B) Inserción de marcapaso bicameral.
C) Ablación septal miocárdica vía percutánea transluminal.
D) Cirugía: miectomía septal, resección quirúrgica del músculo obstructivo más reemplazo o plicación de válvula mitral, transplante cardíaco.
Los resultados a largo plazo de todas estas opciones de tratamiento no están bien definidas.
A) Terapia médica: Esta es la única opción para todos aquellos pacientes que no tienen obstrucción, que son la mayoría. Los beta bloqueadores han sido el tratamiento fundamental en ésta condición; con su uso mejoran la angina, la disnea y el presíncope. También pueden evitar el aumento de gradiente que ocurre con ejercicio al reducir la respuesta inotrópica, en aquellos individuos que tienen obstrucción; aunque el gradiente de presión en reposo se mantiene casi sin cambios. En los niños los beta bloqueadores a altas dosis (5 a 23 mg/k/día), mejoran la sobrevida (dosis convencional de 0.8 a 4mg/k/día). El verapamilo también se ha usado intensamente. Los bloqueadores de los canales de calcio del tipo verapamilo, ejercen una efecto beneficioso en síntomas clínicos como la angina y en la sobrevida de los pacientes con MH. Los efectos del verapamilo se han atribuido predominantemente a que mejoran el llenado diastólico temprano en forma persistente aún después de tratamiento por largo tiempo. Puesto que el volumen sistólico en esta condición es casi fijo, el gasto cardíaco depende mucho de la frecuencia cardíaca. Por esta razón, cada paciente necesita recibir la dosis apropiada de medicación para alcanzar la frecuencia cardíaca óptima para su estado fisiológico. La disopiramida puede ser útil para atenuar el gradiente de presión intraventricular.
B) El marcapaso bicameral provoca mejoría de los síntomas e índices hemodinámicos durante los primeros meses de puesto; y aún un año después. El marcapaso DDD está asociado con un pronóstico excelente en un subgrupo de pacientes severamente comprometidos, muchos de los cuales sufren de síncope o presíncope. No se requieren estudios de marcapaso de base para identificar los pacientes quienes se beneficiarían potencialmente con éste. El bloqueo de rama izquierdo preexistente es compatible con obstrucción severa del (TSVI) y este subgrupo también se beneficia. Además de que disminuye los gradientes de presión en reposo, tanto como los provocables, también se ha visto que se reduce el grosor de la pared en algunos casos. Un estudio randomizado con 48 pacientes sugirió que la mejoría sintomático percibido era consistente con efecto placebo. Esto porque no se demostraron objetivamente los beneficios en cuanto función cardíovascular. Sin embargo en el estudio PIC de la Sociedad Europea de Cardiología, a 80 pacientes se les colocó un marcapaso atrioventricular sincronizado por MH obstructiva refractaria a tratamiento con drogas. Los efectos beneficiosos se demostraron hasta un año después, incluyendo reducción del gradiente de presión en el TSVI. Este fue un estudio cruzado doble ciego con el fin de descartar el efecto placebo. En este estudio hubo dos ramas, en una el marcapaso se dejaba inactivo, y en la otra se programaba activo. Después de tres meses se alternaban ambos modos por tres meses más, y luego se programaba el marcapaso con el modo escogido por el paciente, es decir con el que el paciente se sentía mejor. El marcapaso en modo activo indujo mejoría significativa en la calidad de vida. La discontinuación de la programación después de un período activo llevó al retorno de los síntomas. 66 pacientes de los 80 prefirieron marcapaso en forma activa y 16 solicitaron que se les activara el aparato antes de terminar el período inactivo. Luego de 6 meses de marcapaso los pacientes sintieron mejoría progresiva sobre la ya existente. Ningún estudio controlado sin embargo ha demostrando un efecto favorable en la sobrevida.
C) La técnica de reducción miocárdica a través de cateterización podría reemplazar la cirugía en algunos pacientes. El objetivo de esta técnica es la destrucción selectiva de la parte hipertrofiada del lado izquierdo del tabique interventricular. Se obstruye temporalmente la primera arteria septal y sí esto reduce el gradiente de presión, entonces se inyecta alcohol a través de un catéter con el balón inflado para producir un infarto localizado. La reducción septal no quirúrgica se asocia con una caída en la obstrucción del TSVI y con un aumento del diámetro de ésta área. La técnica también resulta en una alteración consistente de la activación septal con íncoordinación secundaria. Esto último podría jugar un rol importante en la reducción del gradiente y en mejoría sintomático como la que ocurre con la inserción de marcapaso DDD. La ablación septal percutánea transluminal con oclusión de ramas septales inducida por alcohol resultó en reducción del gradiente de presión en el TSVI en varios grupos de pacientes. Esta es una nueva terapia que resulta en reducción del gradiente intraventricular inmediatamente después o poco tiempo después del procedimiento, con resolución de los síntomas. Lakkis de la Universidad de Baylor publica una serie de 50 pacientes a los que se les realiza reducción septal no quirúrgica en los cuales persisten los buenos resultados un año después. Estos resultados se han medido con métodos objetivos como el Doppler, donde se demuestra mejoría en el llenado ventricular izquierdo y la función atrial izquierda. A un grupo de 114 pacientes la mayoría en clase III NYHA se les efectúo reducción septal no quirúrgica. En los primeros 30 pacientes se ocluyeron de una a tres ramas septales con injección de 3.4 +/- 1.6 ml de alcohol absoluto, vía lumen central después de oclusión con balón de la parte proximal de la rama septal. En el resto se realizó ecocardiografía de contraste, de manera que solo una rama se ocluyó. Se alcanzó reducción en el gradiente en reposo en el TSVI en 107 pacientes (94%) de 73.8 +/- 36.5 a 18.6 +/- 19.7 mmHg (p < 0.00001). El aumento máximo en elevación de CK fue 647 +/-330 U/L. Hubo 2 muertes (1.8%) durante la estadía hospitalaria. En 11 pacientes se presentó bloqueo trifascicular permanente (9.6%) y requirieron marcapaso permanente. Control posterior mostró mejoría sintomático sin complicaciones. La visualización del área de ablación previo a la inducción de necrosis química es posible utilizando ecocardiografía de contraste miocárdico intraprocedimiento. Hay un reporte de dos pacientes en quienes el eco de contraste mostró opacificación del músculo papilar medial o la pared libre posterolateral. En ambos pacientes se pudo identificar el área correcta de ablación, al cambiar el vaso septal a ocluir evitando así errores potencialmente fatales. En otro grupo de 62 ablaciones transcoronarias por hipertrofia septal, con inyección de 4.6 ml. de etanol al 96% en las ramas septales en 50 pacientes con MH obstructiva y síntomas severos; la ablación transcoronaria llevó a una reducción en el grosor septal, a eliminación sostenida de la obstrucción (51+/-41 vs 6+/-10 mmHg en reposo, P<0.001; 134+/-48 vs 28+/-32 mmHg, P<0.001, post-extrasistólica), a una disminución en la presión de llenado del VI en reposo y durante ejercicio y a una pronunciada mejoría clínica. No hubo evidencia de creación de un sustrato arritmogénico al evaluarse seriadamente con estimulación eléctrica programada en 39 pacientes. Sin embargo, en el 17% ocurrió bloqueo atrioventricular permanente de alto grado. Hubo dos muertes tempranas, y no hubo muertes tardías durante un seguimiento de 10.6+/-5.6 meses.
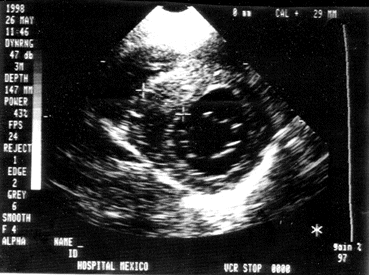
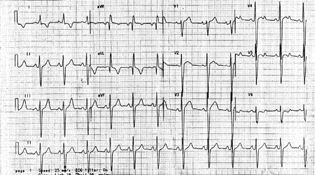
El objetivo de la cirugía es reducir el obstáculo intraventricular, mejorar la compliance del VI, corregir la regurgitación mitral y otras lesiones asociadas, tales como enfermedad arterial coronaria o endocarditis. Se han propuesto varias técnicas quirúrgicas en los últimos 40 años: miotomía, miomectomía, reemplazo de válvula mitral y plicación de la vávula mitral. Esta última combinada con miomectomía puede ser hecha en forma segura y es una alternativa al reemplazo en casos de vávula mitral con valvas grandes y elongadas. La miectomía septal es el procedimiento quirúrgico más frecuentemente efectuado en MH con obstrucción en el tracto de salida del VI. La miectomía septal o resección quirúrgica del músculo obstructivo, reduce o elimina el gradiente en el tracto de salida, y provee mejoría sintomático en la mayoría de los pacientes. El estado clínico postoperatorio puede ser afectado por arritmias y disfunción miocárdica. La figura 1, muestra una imagen del ecocardiograma bidimensional en eje corto, donde se observa el VI a nivel basal de un paciente de 26 años del Hospital México, a quién se le efectuó el procedimiento de Morrow en los Estados Unidos hace 7 años, el paciente sufría de obstrucción severa. Su evolución posterior a sido muy buena pero ocasionalmente presenta angina de pecho generalmente relacionado con la suspensión de medicamentos, verapamilo o beta bloqueadores. La flecha señala el sitio de la cirugía, y como se puede observar la hipertrofia es severa y compromete tanto el tabique anterior como el posterior. La figura 2 muestra el ECG de este paciente. En un estudio no randomizado que comparó miectomía quirúrgica con marcapaso de doble cámara en pacientes con MH obstructiva sintomático, se encontró que la miectomía ofreció más reducción en el gradiente en el tracto de salida del VI y más mejoría subjetiva y en mediciones objetivas de capacidad funcional que el marcapaso bicameral. De un grupo de 519 pacientes operados con míectomía transaórtica subvalvular en MH obstructiva desde 1963; la mayoría de los pacientes estaban en insuficiencia cardíaca NYHA clase III, y llegaron a cirugía después de tratamiento médico por largo tiempo, finalmente insuficiente. El riesgo perioperatorio pudo ser reducido considerablemente durante los años recientes, a pesar de un estado miocardiopático avanzado. La observación postoperatoria a largo plazo demostró unos resultados inesperadamente buenos. Estos resultados deben servir como un estándar a comparar con los obtenidos con la ablación transcoronaria con alcohol. En 66 pacientes la miotomía-miomectomía, miotomía-miomectomía y reemplazo o reparación de la válvula mitral la mortalidad a 30 días fue del 7.5%. También se efectúa, en lugar de reemplazo de la válvula una extensión de la válva anterior de la mitral y miectomía. La extensión de la valva se hace con un parche autólogo de pericardio preservado en glutaraldehído y se usa para alargar la válvula mitral a lo largo de su eje horizontal. Este se considera un abordaje quirúrgico nuevo en MH obstructiva que podría llegar a ser superior a la míectomía sola. Se necesitan más estudios para determinar el valor clínico de este procedimiento. En niños la MH sintomático usualmente tienen un pronóstico grave y debe dársele consideración para trasplante cardíaco tempranamente. En el futuro cercano se podrán reconocer los genotipos con cursos más malignos que se podrán manejar agresivamente para evitar muerte súbita. El problema más grande es como identificar estos pacientes que estarían en riesgo de muerte súbita al afectuar actividades atléticas. Al momento actual la MH familiar presenta un problema extremo de manejo médico. El conocimiento reciente de las bases genéticas de la MH no ha llevado todavía a nuevos tratamientos, porque la capacidad diagnóstica ha sobrepasado las opciones terapéuticas. Una de las cosas más importantes y a la vez la más difícil es la identificación de los pacientes que están en alto riesgo de muerte súbita y requieren terapia agresiva.
Arrítmias y muerte súbita.
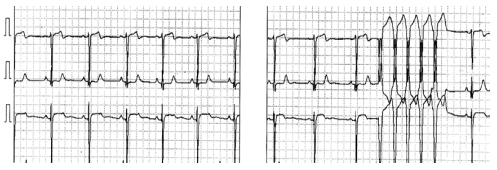
La fibrilación atrial es una causa común de palpitaciones en este trastorno el cual también se asocia a taquicardia ventricular. Tanto la fibrilación atrial como la taquicardia ventricular pueden cambiar mucho las manifestaciones de la MH y pueden ser causa de síncope. También se pueden presentar el fluter atrial y taquicardias supraventriculares. En MH el tamaño del VI (pequeño) parece tener importancia fisiopatológica en el desarrollo de fibrilación atrial. La taquicardia supraventricular causa marcado deterioro hemodinámico que se puede relacionar con historia de síncope, hipertrofia marcada, hiperquinesia, pequeña cavidad ventricular, con volumen de llenado reducido. Puesto que el accidente tromboembólico es el factor más importante de mortalidad y morbilidad asociado con fíbrilación atrial, estos pacientes deben estar anticoagulados. Hay un caso reportado de un paciente con MH quién desarrolló fibrilación atrial sintomático en dos ocasiones después de ingerir citrato de sildenafil (Viagra). El citrato de Sildenafil debe evitarse o usarse con mucha precaución en personas con MH. La figura 3 muestra un trazo de un monitoreo continuo de 24 horas, de un paciente de 46 años portador de MH obstructiva sintomático, con un gradiente intraventricular en reposo de 40mmHg. Este paciente es controlado en la consulta externa del Hospital México, y sus síntomas principales son disnea y síncope no relacionado con esfuerzo. En el ECG se observa una taquicardia ventricular no sostenida o autolimitada.
Conclusión
La MH es una enfermedad familiar del músculo cardíaco causado por anormalidades de los genes de las proteínas contráctiles del sarcómero. Hasta el momento se han reconocido nueve de ellas. La condición se ha definido clínica y patológicamente como un síndrome de hipertrofia miocárdica inexplicada, la cual se asocia con características fisiopatológicas (disfunción diastólica, isquemia, respuestas vasculares alteradas) y patológicas (desorganización de miocitos, aumento del tejido conectivo, anormalidades de los pequeños vasos). La expresión de la enfermedad es variable. Observaciones recientes sugieren que la marcada heterogeneidad clínica y patológica, lo cual ha causado controversia en MH por largo tiempo, es por lo menos en parte función de los genes causantes de la enfermedad. Al momento actual la MH familiar presenta un problema extremo de manejo médico. El conocimiento reciente de las bases genéticas de la MH no ha llevado todavía a nuevos tratamientos. La capacidad diagnóstica ha sobrepasado las opciones terapéuticas. La taquicardia ventricular y la fibrilación ventricular son el principal mecanismo de muerte súbita en la MH. En pacientes de alto riesgo, los cardiodesfibriladores implantables son altamente efectivos para terminar tales arritmias, lo que indica que estos aparatos juegan un rol en prevención primaria y secundaria de muerte súbita. En estos pacientes en los que se sospecha un alto riesgo de muerte súbita arrítmica el único tratamiento efectivo al momento es el desfibrilador implantable.
Referencias
1.Roberts R. A perspective: the new millennium dawns on a new paradigm for cardiology molecular genetics. J Am Coll Cardiol 2000;36:661-667 [ Links ]
2.Spirito P, Seidman C E, McKenna W J, Maron B J. The management of hypertrophic cardiomyopathy. N Eng J Med 1997;336:775-785 [ Links ]
3.Goodwin JF. Cardiomyopathies and Specific Heart Muscle Diseases. Definitions, Terminology, Classifications and New and Old Approaches. Postgrad Med-J 1992; 68 Suppl 1: S3-6 [ Links ]
4.Matsubara T, Yamazoe M, Kimura M, Hori T, Ozaki K, Hatada K, Aizawa Y. Hypertrophic cardiomyopathy with dominant hypertrophy in the right anterobasal region of the ventricular septum: a case report. J Cardiol 2000;36:45-48 [ Links ]
5.Maron BJ; Gardin JM; Flack JM; Gidding SS; Kurosaki TT; Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92: 785-789. [ Links ]
6.Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 22;102:858-864 [ Links ]
7.Murgo JP, Alter BR, Dorethy JF, Altobelli SA, Mc Granahan GM. Dynamics of left ventricular eyection in obstructive and non obstructive hypertrofic cardiomyopathy. J Clin Invest 1980;68:1369-1382 [ Links ]
8.Vlachopoulos C, ORourke M. Genesis of the Normal and Abnormal Arterial Pulse. Current Problems in Cardiology 2000;25:5.348 [ Links ]
9.Zimetbaum P, Josephson M. Evaluation of Patients with Palpitations. N Eng J Med 1998;338: 1371 [ Links ]
10.Maron B J, Moller J, Seidman C E, Vincent M, Dietz H C, Moss A J. Impact of Laboratory Molecular Diagnosis on Contemporary Diagnostic Criteria for Genetically Transmitted Cardiovascular Disease: Hypetrophic Cardiomiopathy, long-QT Syndrome, and Marphan Syndrome. A Statement for Helthcare Professionals from the Councils on Clinical Cardiology, Cardiovascular Disease in the Young, and Basic Science, American Heart Association. Circulation 1998;98:1460-1471 [ Links ]
11.Fukuda N; Oki T; Iuchi A; Tabata T; Manabe K; Kageji Y; et al. Clin Cardiol 1996; 19: 121-127 [ Links ]
12.Baracca E; Brunazzi MC; Pasqualini M; Cavazzini D; Scorzoni D; Vaccari et al. Ventricular diastolic function from the spectral analysis of the fourth heart sound. A Doppler validated study in hypertrophic cardiomyopathy. Acta-Cardiol 1995; 50: 17-21 [ Links ]
13.Briguori C, Betocchi S, Romano M, Manganelli F, Angela Losi M, Ciampi Q, et al. Exercise capacity in hypertrophic cardiomyopathy depends on left ventricular diastolic function. Am J Cardiol 1999; 84:309-315 [ Links ]
14.Marian AJ. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet 2000;355:58-60 [ Links ]
15.Moro E; D'Angelo G; Nicolosi GL; Mimo R; Zanuttini. Apical hypertrophic cardiomyopathy. Eur Heart J. 1995; 16: 210-217 [ Links ]
16.Leachman RD. Potential mechanisms of improvement after various treatments for hypertrophic obstructive cardiomyopathy.Tex-Heart-Inst-J 1995; 22: 126-133 [ Links ]
17.Wynne J y Braunwald E. The cardiomyopathies and myocarditides. En: Braunwald (editor). Heart Hisease. A Texbook of Cardiovascular Medicine.WB Saunders Company. 5th edition. (1997); pág:1404-1453 [ Links ]
18.Ostman Smith I, Wettrell G, Riesenfeld T. A cohort study of childhood hypertrophic cardiomyopathy: improved survival following high-dose beta-adrenoceptor antagonist treatment. J Am Coll Cardiol 1999;34:1813-1822 [ Links ]
19.Hartmann A; Schnell J; Hopf R; Kneissl G. Persisting effect of Ca(2+)-channel blockers on left ventricular function in hypertrophic cardiomyopathy after 14 years' treatment. Angiology 1996; 47: 765-773 [ Links ]
20.Tokudome T, Mizushige K, Ueda T, Sakamoto S, Matsuo H. Effect of disopyramide on left ventricular pressure gradient in hypertrophic obstructive cardiomyopathy in comparison with propranolol--a case report. Angiology 1999;50:331-335 [ Links ]
21.Fananapazir L; Epstein ND; Curiel RV; Panza JA; Tripodi D; McAreavey D. Long-term results of dual-chamber (DDD) pacing in obstructive hypertrophic cardiomyopathy. Evidence for progressive symptomatic and hemodynamic improvement and reduction of left ventricular hypertrophy.Circulation 1994; 90: 2731 [ Links ]
22.Maron BJ, Nishimura RA, McKenna WJ, Rakowski H, Josephson ME, Kieval RS Assessment of permanent dual-chamber pacing as a treatment for drug-refractory symptomatic patients with obstructive hypertrophic cardiomyopathy. A randomized, double-blind, crossover study (M-PATHY). Circulation 1999;99:2927-2933 [ Links ]
23.Gadler F, Linde C, Daubert C, McKenna W, Meisel E, Aliot E, et al, Significant improvement of quality of life following atrioventricular synchronous pacing in patients with hypertrophic obstructive cardiomyopathy. Data from 1 year of follow-up. PIC study group. Pacing In Cardiomyopathy. Eur Heart J 1999;20:1044-1050 [ Links ]
24.Sadoul N; Simon JP; de Chillou C; Bruntz JF; Isaaz K; Beurrier D, et al. Value of permanent cardiac pacing in hypertrophic and obstructive cardiomyopathies resistant to medical treatment. Arch Mal Coeur Vaiss 1994; 87: 1315-1323 [ Links ]
25.Sigwart U. Non-surgical myocardial reduction for hypertrophic obstructive cardiomyopathy. Lancet 1995; 346: 211-214 [ Links ]
26.Henein MY, O'Sullivan CA, Ramzy IS, Sigwart U, Gibson DG. Electromechanical left ventricular behavior after nonsurgical septal reduction in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol 1999;34:1117-1122 [ Links ]
27.Lakkis NM, Nagueh SF, Dunn JK, Killip D, Spencer WH 3rd. Nonsurgical septal reduction therapy for hypertrophic obstructive cardiomyopathy: one-year follow-up. J Am Coll Cardiol 2000;36:852-855 [ Links ]
28.Nagueh SF, Lakkis NM, Middleton KJ, Killip D, Zoghbi WA, Quiñones MA, Spencer WH 3rd. Changes in left ventricular filling and left atrial function six months after nonsurgical septal reduction therapy for hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol 1999;34:1123 [ Links ]
29.Seggewiss H, Faber L, Gleichmann U. Percutaneous transluminal septal ablation in hypertrophic obstructive cardiomyopathy. Thorac Cardiovasc Surg 1999;47:94-100 [ Links ]
30.Faber L, Seggewiss H, Ziemssen P, Gleichmann U. Intraprocedural myocardial contrast echocardiography as a routine procedure in percutaneous transluminal septal myocardial ablation: detection of threatening myocardial necrosis distant from the septal target area. Cardiovasc Interv Catheter 1999;47:462-466 [ Links ]
31.Gietzen FH, Leuner CJ, Raute-Kreinsen U, Dellmann A, Hegselmann J, Strunk-Mueller C, Kuhn HJ. Acute and long-term results after transcoronary ablation of septal hypertrophy (TASH). Catheter interventional treatment for hypertrophic obstructive cardiomyopathy. Eur Heart J 1999;20:1342-1354 [ Links ]
32.Herreros J; Fulquet E; Florez S; Echevarria R. Surgical treatment of hypertrophic obstructive myocardiopathy: is it an underestimated alternative? Rev Esp Cardiol 1996; 49: 372-380 [ Links ]
33.Maron BJ, Merril WH, Freir PA, Kent KM, Epstein SE,Morrow AG. Long term clinical course and symptomatic statusof patients after the operations for hypertrophic subaortic stenosis. Circulation 1978;57:1205-1213 [ Links ]
34.Merrill WH, Friesinger GC, Graham TP Jr, Byrd BF 3rd, Drinkwater DC Jr, Christian KG, Bender HW Jr. Long-lasting improvement after septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2000;69:1732-1735 [ Links ]
35.Ommen SR, Nishimura RA, Squires RW, Schaff HV, Danielson GK, Tajik AJ Comparison of dual-chamber pacing versus septal myectomy for the treatment of patients with hypertropic obstructive cardiomyopathy: a comparison of objective hemodynamic and exercise end points. J Am Coll Cardiol 1999;34:191-196 [ Links ]
36.Schulte HD, Borisov K, Gams E, Gramsch-Zabel H, Losse B, Schwartzkopff B. Management of symptomatic hypertrophic obstructive cardiomyopathy-long-term results after surgical therapy.Thorac Cardiovasc Surg 1999;47:213-218 [ Links ]
37.Jault F; Gandjbakhch I; Rama A; Nataf P; Dorent R; Bors V;et al. Long term results of the surgical treatment of obstructive hypertrophic. Arch Mal Coeur Vaiss 1996; 89: 679-684 [ Links ]
38.Kofflard MJ; van Herwerden LA; Waldstein DJ; Ruygrok P; Boersma E; Taams MA; et al. J Am Coll Cardiol 1996; 28: 197-202 [ Links ]
39.Bryant RM. Hypertrophic cardiomyopathy in children. Cardiol Rev 1999;7:92-100 [ Links ]
40.Leachman RD. Potential mechanisms of improvement after various treatments for hypertrophic obstructive cardiomyopathy. Tex Heart Inst J 1995; 22: 126-133 [ Links ]
41.McKenna WJ. The future in hypertrophic cardiomyopathy: important clues and potential advances from an understanding of the genotype phenotype relationship. Ital Heart J 2000 Jan;1:17-20 [ Links ]
42.Zimetbaum P, Josephson M. Evaluation of Patients with Palpitations.N Eng J Med 1998;338:19 [ Links ]
43.Nakatani M; Yokota Y; Yokoyama Clin Cardiol. 1996; 19: 385-392 [ Links ]
44.Manganelli F, Betocchi S, Losi MA, Briguori C, Pace L, Ciampi Q, et al Influence of left ventricular cavity size on clinical presentation in hypertrophic cardiomyopathy. Am J Cardiol 1999 15;83:547-552 [ Links ]
45.Awan GM, Calderon E, Dawood G, Alpert MA Acute, symptomatic atrial fibrillation after sildenafil citrate therapy in a patient with hypertrophic obstructive cardiomyopathy Am J Med Sci 2000;320:69-71 [ Links ]
46.Khan IA, Ajatta FO, Ansari AW. Persistent ST segment elevation: a new ECG finding in hypertrophic cardiomyopathy. Am J Emerg Med 1999;17:296-299 [ Links ]
47.Candell Riera J. Role of noninvasive techniques (electrocardiogram, Holter, tilt-table test, nuclear magnetic resonance, isotopes) in the evaluation of patients with hypertrophic cardiomyopathies. Rev Esp Cardiol 1995; 48: 828-836 [ Links ]
48.Shimizu M, Ino H, Okeie K, Emoto Y, Yamaguchi M, Yasuda T,et al.Exercise-induced ST-segment depression and systolic dysfunction in patients with nonobstructive hypertrophic cardiomyopathy Am Heart J 2000;140:52-60 [ Links ]
49.Dritsas A; Sbarouni E; Gilligan D; Nihoyannopoulos P; Oakley CM. QT-interval abnormalities in hypertrophic cardiomyopathy. Clin Cardiol 1992;15: 739-742 [ Links ]
50.Peters S, Rust H, Trummel M, Brattstrom A Familial hypertrophic cardiomyopathy associated with prolongation of the QT interval. Z Kardiol 2000;89:624-629 [ Links ]
51.MacRae CA; Ghaisas N; Kass S; Donnelly S; Basson CT; Watkins HC. Familial Hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome maps to a locus on chromosome 7q3.J-Clin-Invest. 1995; 96: 1216-1220 [ Links ]
52.Gonzalez-Torrecilla E; Garcia-Fernandez MA; Bueno H; San Roman D; Moreno MM; Bermejo J; et al The mitral valve apparatus in hypertrophic myocardiopathy: observations by transesophageal echocardiography Rev Esp Cardiol. 1995; 48: 542-551 [ Links ]
53.Torrecilla EG, Garcia Fernandez MA, Bueno H, Moreno M, Delcan JL Pulmonary venous flow in hypertrophic cardiomyopathy as assessed by the transoesophageal approach. The additive value of pulmonary venous flow and left atrial size variables in estimating the mitral inflow pattern in hypertrophic cardiomyopathy [ Links ]
53. Pai RG, Stoletniy LN An integrated measure of left ventricular diastolic function based on relative rates of mitral E and A wave propagation. J Am Soc Echocardiogr 1999;12:811-816 [ Links ]
54.Nagueh SF, Lakkis NM, Middleton KJ, Spencer WH 3rd, Zoghbi WA, Quinones MA. Doppler estimation of left ventricular filling pressures in patients with hypertrophic cardiomyopathy. Circulation 1999 19;99:254-261 [ Links ]
55.Nakamura T, Sugihara H, Kinoshita N, Yoneyama S, Azuma A, Nakagawa M. Can serum carnitine levels distinguish hypertrophic cardiomyopathy from hypertensive hearts? Hypertension 2000;36:215-219 [ Links ]
56.Sharma S, Elliott PM, Whyte G, Mahon N, Virdee MS, Mist B,et al. Utility of metabolic exercise testing in distinguishing hypertrophic cardiomyopathy from physiologic left ventricular hypertrophy in athletes. J Am Coll Cardiol 2000;36:864-870 [ Links ]
57.Lazzeroni E; Picano E; Dodi C; MorozziL; Chiriatti GP; Lu C; et al. Dipyridamole echocardiography for diagnosis of coexistent coronary artery disease in hypertrophic cardiomyopathy. Echo-Persantine International Cooperative (EPIC) Study Group-Subproject Hypertrophic Cardiomyopathy. Am-J-Cardiol. 1995 15; 75: 810-813 [ Links ]
58.Okeie K, Shimizu M, Yoshio H, Ino H, Yamaguchi M, Matsuyama T, et al. Left ventricular systolic dysfunction during exercise and dobutamine stress in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2000;36:856-863 [ Links ]
59.Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun 1999;262:411-417 [ Links ]
60.Bundgaard H, Havndrup O, Andersen PS, Larsen LA, Brandt NJ, Vuust J, et al. Familial hypertrophic cardiomyopathy associated with a novel missense mutation affecting the ATP-binding region of the cardiac beta-myosin heavy chain. J Biol Chem 2000;275:28045-52 [ Links ]
61.Yamashita H, Tyska MJ, Warshaw DM, Lowey S, Trybus KM Muraishi A, et al. Malalignment of the sarcomeric filaments in hypertrophic c-ardiomyopathy with cardiac myosin heavy chain gene mutation. Heart 1999;82:625-629 [ Links ]
62.Mukherjea P, Tong L, Seidman JG, Seidman CE, Hitchcock-DeGregori SE. Altered regulatory function of two familial hypertrophic cardiomyopathy troponin T mutants. Biochemistry 1999;38:13296-30 [ Links ]
63.Varnava A, Baboonian C, Davison F, de Cruz L, Elliott PM, Davies MJ, et al. A new mutation of the cardiac troponin T gene causing familial hypertrophic cardiomyopathy without left ventricular hypertrophy. Heart 1999;82:621-624 [ Links ]
64.Towbin JA. Molecular Genetics of Hypertrophic Cardiomyopathy. Curr Cardiol Rep 2000;2:134-140 [ Links ]
65.Gruen M, Gautel M Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol 1999;286(3):933-949 [ Links ]
66.Doi YL, Kitaoka H, Hitomi N, Satoh M, Kimura A. Clinical expression in patients with hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutation. Circulation 1999;100:448-449 [ Links ]
67.Morano I. Tuning the human heart molecular motors by myosin light chains. J Mol Med 1999;77:544-555 [ Links ]
68.Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, et al. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest 1999;103:R39-43 [ Links ]
69.Watkins H Rosenzweig A, Hwang D S, et al. Characteristics and prognostic implications of myosine missense mutations in familial hypertrofic cardiomyopathy. N Eng J Med 1992;326:1108-1114 [ Links ]
70.Watkins H, McKenna WJ, Thierfelder L, et al. Mutations in the genes for cardiac troponin T and alfa tropomyosin in hypetrophic cardiomyopathy. N Eng J Med 1995;332:1068-1064 [ Links ]
71.Niimura H, Bachinski LL, Sangwataronaroj S, et al. Mutations in the gene for cardiac myosin-protein C and late onset familial hypertrophic cardiomyopathy.N Engl J Med 1998;82:621-624 [ Links ]
72.Teare D, Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1-18 [ Links ]
73.Dieckerhoff J; Frenzel H; Schulte HD; Losse B; Motz W: Dysplastic intramyocardial arteries with subaortic septum in patients with hypertrophic obstructive cardiomyopathy. Z Kardiol 1992; 81: 456-463 [ Links ]
74.Davies MJ; McKenna WJ. Hypertrophic cardiomyopathy--pathology and pathogenesis. Histopathology. 1995; 26: 493-500 [ Links ]
75.Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A. Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol 2000;31:988-998 [ Links ]
76.Davies MJ.The investigation of sudden cardiac death. Histopathology 1999;34:93-98 [ Links ]
77.Brugada J. Sudden death in hypertrophic myocardiopathy. Rev Esp Cardiol 1998;51:991-996 [ Links ]
78.Watkins H. Sudden Death in Hypertrophic Cardiomyopathy The N Engl J Med 2000; 342, 422-424 [ Links ]
79.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden eath in hypertrophic cardiomyopathy N Engl J Med 2000;342:1778-1785 [ Links ]
80.Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy.J Am Coll Cardiol 999;33:2044-2051 [ Links ]
81.Zhang R, Zhao J, Jones M, Guzman G, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. J Biol Chem 2000;275:624 [ Links ]
82.Mukherjea P, Tong L, Seidman JG, Seidman CE, Hitchcock-DeGregori SE. Altered regulatory function of two familial hypertrophic cardiomyopathy troponin T mutants. Biochemistry 1999;38(40):13296-13301 [ Links ]
83.Varnava A, Baboonian C, Davison F, de Cruz L, Elliott PM, Davies MJ, McKenna WJ A new mutation of the cardiac troponin T gene causing familial hypertrophic cardiomyopathy without left ventricular hypertrophy. Heart 1999;82:621-624 [ Links ]
84.Maron B J, Win Kuang Shen, Link MS., Epstein AE, Almquist AK, Daubert JP, et al. Efficacy of Implantable Cardioverter-Defibrillators for the Prevention of Sudden Death in Patients with Hypertrophic Cardiomyopathy. N Engl J Med 2000;342:365-373 [ Links ]
85.Mckenna WJ, Oakley CM, Krikler DM, Goodwin JF. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart Journal 1985;53:416-426 [ Links ]
86.Elliott PM, Sharma S, Varnava A, Poloniecki J, Rowland E, McKenna WJ. Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999;33:1596-601 [ Links ]
87.The Antiarrhythmics versus implantable defibrilators (AVID) investigators. A comparison of antiarrythmic-drug therapy with implantable defribrilators in patients resuscitated from near fatal ventricular arrhyhmias.N Engl J med 1997;337:1576-1583 [ Links ]
88.Sra J, Dhala A, Blanck Z, Deshpande S, Cooley R, Akhtar M. Sudden cardiac death. Curr prob cardiol 1999;24:461-538. [ Links ]
89.Tabib A, Miras A, Taniere P, Loire R. Undetected cardiac lesions cause unexpected sudden cardiac death during occasional sport activity. A report of 80 cases. Eur Heart J 1999;20:900-903 [ Links ]
90.Kurowski K, Chandran S The preparticipation athletic evaluation. Am Fam Physician 2000;61:2683-90, 2696-2698 [ Links ]
91.Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998 6;339:364-369. [ Links ]
92.McKenna WJ. The future in hypertrophic cardiomyopathy: important clues and potential advances from an understanding of the genotype phenotype relationship. Ital Heart J 2000;1:17-20 [ Links ]
93.DeLuca M, Tak T. Hypertrophic cardiomyopathy. Tools for identifying risk and alleviating symptoms. Postgrad Med 2000;107:127-130, 133-5, 139-40 [ Links ]
*Asistente del Servicio de Cardiología del Hospital México, CCSS, San José, Costa Rica.
Instructora de las Cátedras de Fisiopatología y Medicina Interna de la Universidad de Costa Rica.
Apartado 172- San José 1017
e-mal:baltesq@ sol. racsa.co.cr

