










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa Pediátrica Costarricense
Print version ISSN 1409-0090
Acta pediátr. costarric vol.17 n.2 San José Jan. 2003
Paula Buitrago Mata (1), Loretta Giacomin Carmiol (2), Annette Vallejo Serrano (3), Silvia Santamaría (4)
(1) Médico.cirujano, Clínica Dr. Carlos Durán Cartín
(2) Médico-cirujano, Servicio de Ginecología del Hospital Dr. Max Peralta.
(3) Médico-cirujano, EBAIS de Monterrey en el Area de Salud de Aserrí.
(4) Médico-cirujano, Patóloga, Servicio de Patología del Hospital Nacional de Niños "Dr. Carlos Saénz Herrera" .
Dirección para correspondencia: Tres Ríos, del Palí 100 metros Este y 75 metros Norte, Teléfono: Celular: 374-6770; Casa: 278-4154; Fax: 279-5428; apartado: 300-2250 Cartago Email: p_buitrago@hotmail.com
Acta Pediátrica Costarricense 2003, volumen 17, número 2.
La enfermedad de Wolman es una lipidosis congénita, que se transmite de una forma autosómica recesiva y se debe al déficit de una enzima lisosómica que es codificada en el brazo largo del cromosoma 10: la lipasa ácida.
El inicio clínico de la enfermedad tiene lugar durante las primeras semanas de vida y se caracteriza por vómitos, diarrea, esteatorrea, hepatoesplenomegalia, distensión abdominal, desnutrición progresiva y detención de la curva ponderal. Tiene una evolución rápidamente fatal y la muerte se produce invariablemente a lo largo del primer año de vida. No hay datos clínicos de rutina que sugieran su diagnóstico definitivo y aunque las calcificaciones bilaterales de las glándulas suprarrenales se consideran patognomónicas, este signo radiológico no está siempre presente.
Para confirmar su diagnóstico es posible determinar la actividad de la lipasa ácida lisosomal en cultivo de fibroblastos o en los leucocitos periféricos del paciente así como en las células del líquido amniótico entre las semanas 16 y 20 de gestación o en cultivo de vellosidades coriónicas .
Aunque actualmente no se dispone de un tratamiento específico y eficaz, es interesante su conocimiento para establecer un adecuado consejo genético, e incluso la posibilidad de un estudio antenatal.
Palabras clave: Enfermedad de Wolman, Enfermedad de depósito de ésteres de colesterol (CESO), calcificaciones suprarrenales, lipasa ácida lisosomal, lipidosis.
Caso clínico
Paciente masculino nacido el 5 de Febrero del 2002 en el Hospital San Juan de Dios, hijo de padres consanguíneos (primos hermanos). Madre de 38 años, conocida sana, G5P4A 1 C1, portadora de Diabetes Mellitus Gestacional y padre sin patología aparente; los hermanos están sanos.
Embarazo con control prenatal adecuado. Parto por cesárea electiva a las 41 semanas de gestación. RNTGEG. Apgar 8-9. Peso al nacer 4000 g, talla 54 cm, CC 37 cm. Examen físico normal.
Captación temprana del neonato en el primer nivel de atención. Referido a los 2 meses de edad a la Clínica Moreno Cañas por cuadro que se manifestó a los 30 días de nacido caracterizado por vómitos de contenido alimentario, distensión abdominal y pérdida de peso.
A la exploración se documentó 4600 g de peso y hepatomegalia. Se interpreta como Reflujo Gastroesofágico, se instaura tratamiento con metoclopramida y leche de soya y se indican estudios (esofagograma, radiografía y US de abdomen). Sin embargo; ante la persistencia de la clínica descrita, es llevado un mes después por sus padres al Hospital Nacional de Niños. Al momento de su ingreso pesaba 4100 g y medía 59.5 cm. El examen físico reveló desnutrición, distensión abdominal y hepatomegalia 4-5 cms bajo el reborde costal derecho. Se descartó cualquier manejo quirúrgico y se ingresó al niño para su estudio. Entre los exámenes de laboratorio y gabinete se destacaron:
Hb: 9.4 g/dl Hto: 27.2%.
Leucocitos: 13.490
EGO: normal
Estudio hormonal suprarrenal normal
Estudios por TORCH: negativos
Grasa en heces ++
TGO: 179 u/L
TP: 30 %, TPT:47 seg, Fibrinógeno: 117
Colesterol: 125 mg/dl, Triglicéridos: 214 mg/dl
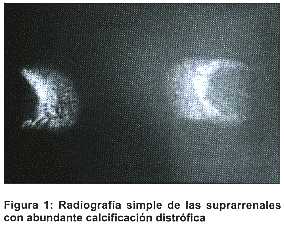
Radiografía de abdomen: distensión abdominal, edema de asas y calcificaciones en glándulas suprarrenales (Figura 1).
US de abdomen: "Hepatomegalia homogénea, glándula suprarrenal derecha calcificada y probable izquierda. Marcada distensión abdominal y asas con edema de pared".
Estudio histológico de Médula Osea: trombopoyesis disminuída, hiperplasia histiocítica con abundantes células espumosas compatible con enfermedad de depósito.
La evolución del paciente fue tórpida; presentó cuadros de diarrea que cedieron parcialmente con hidratación, picos febriles sin respuesta a antibiótico terapia, pérdida progresiva de peso y empeoramiento de su anemia y coagulopatía con aumento progresivo del TP y TPT y disminución del recuento plaquetario y del fibinógeno.
Falleció 14 días después de su ingreso.
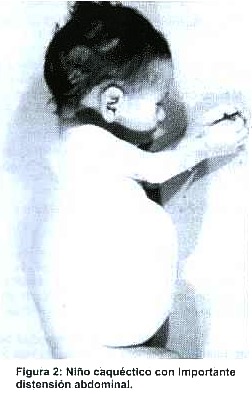
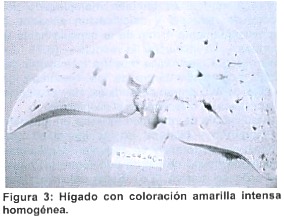
En la autopsia se encontró un niño marasmático, con distensión abdominal severa (Figura 2). El hígado tenía marcada coloración amarilla y estaba ligeramente aumentado de consistencia (Figura 3). En el examen histológico mostraba hepatocitos y células de Kupffer aumentados de tamaño, vacuolados y presencia de abundantes macrófagos espumosos en el espacio portal y periportal con borramiento del patrón lobulillar. Había necrosis de las células espumosas y hepatocitos portales y periportales. Además se observaron abundantes cristales birrefringentes dentro de los macrófagos, hepatocitos y células de Kupffer.
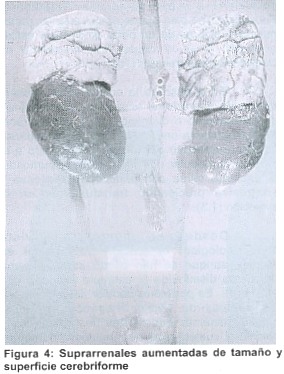
Las suprarrenales estaban muy aumentadas de tamaño con peso de 26 g (Figura 4) y mostraban capa glomerular conservada; la fascicular interna y la reticular ensanchadas por macrófagos espumosos, muchos de ellos necróticos que contenían cristales birrefringentes. Había abundante calcificación distrófica. El tejido linfoide y la médula ósea mostraban abundantes macrófagos que sustituían a las células parenquimatosas. El intestino delgado tenía coloración amarillenta en su superficie mucosa y abundantes histiocitos espumosos con cristales en la lámina propia y las vellosidades se veían engrosadas y acortadas. La microscopía electrónica mostraba depósito de lípidos y cristales intralisosomales en hígado, suprarrenales, intestino, bazo, médula ósea, pulmones, ganglios linfáticos, bazo, testículos, tiroides, timo y amígdalas.
Discusión
La enfermedad de Wolman es una alteración congénita del metabolismo de los lípidos y se debe al déficit de una enzima lisosómica: la lipasa ácida -EC 3.1.1.13 - (1-14)
Esta enzima - específicamente su isoenzima A - (3,11,14) es codificada en el brazo largo del cromosoma 10 (1-3,9,14) y su función consiste en la degradación de triglicéridos de cadena corta y larga y ésteres de colesterol a sus unidades básicas arquitecturales; un defecto en su función producirá el acúmulo de estas macromoléculas en la mayoría de los órganos y tejidos.
La enfermedad fue descrita por primera vez por Abramov, Schorr y Wolman en 1956 (1,3, 9,13,14) al objetivar acúmulos de triglicéridos y ésteres de colesterol en el hígado, bazo, glándulas suprarrenales y ganglios linfáticos de un lactante.
En 1969, Patrick y Lake demostraron que este acúmulo era secundario al déficit de la lipasa ácida lisosomal, enzima responsable de la hidrólisis de estos compuestos al pasar de sangre periférica a la célula particularmente cuando éstos forman lipopoteínas de baja densidad (1). El primer caso fue reportado en Israel, luego en Estados Unidos posterior a lo cual ha sido documentada en la mayoría de los países y grupos étnicos. No obstante, parece ser más prominente en los países del oeste y centro de Europa, Arabia Saudita, India, Canadá y otros (7).
La enfermedad se transmite de una forma autosómica recesiva (1, 2, 3, 4, 6, 7, 8, 9, 11, 14), con un reparto equitativo en ambos sexos y frecuentemente con varios casos en la misma familia (3), conformada a veces por padres consaguíneos (2, 7). Sin embargo, es difícil precisar con exactitud su incidencia debido a que los pediatras varían en su habilidad para diagnosticarla (7).
Es importante anotar que el déficit de la lipasa ácida lisosomal tiene dos expresiones fenotípicas y además de la ya mencionada enfermedad de Wolman, existe otro trastorno que representa una variante alélica del mismo locus genético conocida como enfermedad de depósito de ésteres de colesterol (CESO) (1, 6, 9, 11,12, 14). Esta es una forma relativamente benigna, que puede pasar inadvertida hasta la edad adulta (6) y en cuyo caso hay una déficit enzimático del 50% de la actividad normal de la enzima que se reduce a menos del 10% en sujetos con Wolman (7,13). La lipólisis a un pH neutro parece no estar afectada razón por la cual su evolución clínica es menos grave. Entre estos dos extremos se ha reportado un número pequeño de pacientes con presentación intermedia. Además se han documentado pacientes con deficiencia de lipasa ácida con otras manifestaciones clínicas lo que sugiere que el espectro clínico de la deficiencia de lipasa ácida necesita extenderse (15). Desde su descubrimiento y hasta el año 1993, se han publicado 125 pacientes con niveles bajos de lipasa ácida lisosomal de los cuales 65 corresponden a enfermedad de Wolman, 49 a CESO y 11 a variantes alélicas intermedias (1). En Costa Rica se han reportado 5 casos.
Manifestaciones Clínicas
El inicio clínico de la Enfermedad de Wolman tiene lugar durante las primeras semanas de vida luego de un breve intervalo libre (3,11,13), y no es improbable que los recién nacidos afectos de esta enfermedad nazcan como niños normales (2,7), aunque excepcionalmente puede iniciarse al nacimiento (1 ).
Generalmente, son hospitalizados en los primeros dos meses de vida (7) y la muerte se produce invariablemente a lo largo del primer año - con mayor frecuencia en el primer o segundo trimestres- (1-11), aunque se han citado supervivencias más largas (3).
La mayoría de los pacientes cursan con un cuadro clínico similiar que se caracteriza por vómitos, diarrea, esteatorrea, hepatoesplenomegalia, distensión abdominal, desnutrición progresiva y detención de la curva ponderal (1-10,12-14). La esplenomegalia puede presentarse en uno de cada tres casos, puede haber enfermedad pulmonar crónica, taquipnea, ascitis y linfadenopatías (14). No hay síntomas de alteración específicos del sistema nervioso central; sin embargo, algunos casos se han citado con reflejos osteotendinosos exaltados y clono. El fondo de ojo no revela la mancha rojo cereza característica de otras tesaurismosis (3). Los cambios patológicos más significativos comprenden el depósito de triglicéridos y ésteres de colesterol en las células del sistema fagocítico, en el intestino delgado y la corteza adrenal.
Las células del sistema reticuloendotelial aparecen muy aumentadas de tamaño y vacuoladas; el déficit enzimático les impide librarse de los ésteres de grasa quedando transformadas en células espumosas responsables de la hepatoesplenomegalia y linfadenopatías (7).
El hígado es grande, firme y amarillo. La arquitectura normal puede estar conservada o tan distorsionada que sólo se identifiquen espacios porta. En la mayoría de los casos puede ser evidente la presencia de células espumosas, fibrosis portal e incluso cirrosis. Es visible la acumulación de lípidos en el parénquima hepático y en las células de Kupffer (3,13,14).
Por otro lado, se produce un ensanchamiento y aplanamiento de las vellosidades intestinales causados por la infiltración severa de la lámina propia por histiocitos repletos de lípidos (2,3,7,14). No solamente se ve afectada la mucosa sino también el resto de capas del intestino y las neuronas del plexo mientérico, lo que explica la prominencia de síntomas gastrointestinales en estos pacientes (por muerte neuronal) y su apariencia en el estudio macroscópico en el cual se observa aterciopelado, suave, brillante y amarillo (7).
El interior de las zonas fascicular y reticular de la corteza adrenal es también reemplazado por focos de necrosis y calcificación situados a aproximadamente 1 mm de la cápsula hacia la corteza - sin involucrar la médula - (6, 7). La microscopía electrónica muestra la mayoría del lípido acumulado dentro de los lisosomas los cuales se encuentran distendidos. Hay presencia de cristales de colesterol en el citoplasma.
La radiografía de abdomen muestra un aumento homogéneo y bilateral de las glándulas que conservan su forma y se observan calcificadas siguiendo desde un patrón puntiforme (4) hasta un proceso masivo (3, 6). Estas calcificaciones son bilaterales y muchos autores las consideran patognomónicas (1-11,14), aunque se han citado casos en la literatura sin este signo radiológico (1,3,6).
Se debe realizar diagnóstico diferencial con otras patologías en las cuales pueden observarse calcificaciones simliares, aunque raras y de localización unilateral. Se incluyen en esta categoría la enfermedad granulomatosa, neoplasias (carcinomas, neuroblastomas, ganglioneuromas, feocromocitomas, teratomas, tumores adrenales), tuberculosis y enfermedad de Addison, (1,3,6). La hemorragia adrenal es secundaria a sepsis (Síndrome Waterhouse-Friderichson) o bien puede observarse en recién nacidos después de un parto difícil o de asfixia perinatal, y es la causa más frecuente de clacificaciones adrenales (16).
En el caso de la ecografía se observa una masa suprarrenal ecogénica, piramidal sin desplazamiento o distorsión de la silueta renal (6) y se observa un hígado grande e hipodenso debido al depósito de lípidos (14). Debido a la infiltración medular por macrófagos espumosos, el paciente cursa con pancitopenia intensa y progresiva precisando transfusiones en la evolución (1,3).
Desde el punto de vista endocrinológico, la función suprarrenal es normal, aunque se han citado casos de una respuesta disminuída al estímulo exógeno con ACTH (3). Es posible evidenciar alteraciones en las pruebas de función hepática así como hipoalbuminemia. Los niveles, de triglicéridos y colesterol suelen estar disminuídos o normales, aunque se han publicado casos con aumento de triglicéridos y VLDL (1,3). Los tiempos de coagulación están aumentados. Debido a la carencia de datos clínicos de rutina que sugieran el diagnóstico definitivo (1,3,14), éste se confirma con la determinación de la actividad de la lipasa ácida lisosomal en cultivo de fibroblastos o en los leucocitos periféricos del paciente (1, 3,12,14). Además es posible determinar la actividad de esta enzima en las células del líquido amniótico entre las semanas 16 y 20 de gestación por lo que se puede realizar un diagnóstico prenatal (1-3,7,10-11,14).
Tratamiento
Aunque no se dispone de un tratamiento específico y eficaz (1, 3, 8, 13), se deben iniciar una serie de medidas desde el momento en que se sospecha el diagnóstico con el objetivo de evitar el asiento de lípidos que no puedan ser transportados y metabolizados en el intestino, ya que el depósito progresivo de grasas produce malabsorción responsable del estado de inanición que conlleva a la muerte de estos pacientes (7). Estas medidas incluyen:
1. Suspender la lactancia materna y/o ingesta de fórmulas que contengan triglicéridos y ésteres de colesterol.
2. Proporcionar una nutrición suficiente vía parenteral que incluya todas las vitaminas incluyendo las liposolubles. Respecto a lo anterior, algunos autores opinan que ni siquiera una dieta carente de grasas mejora la diarrea y corrige el síndrome de malabsorción mientras que otros creen que la hiperalimentación parenteral puede mejorar en parte el deterioro nutricional (9). No obstante, aunque el balance calórico pueda mantenerse por este tipo de alimentación, se debe estar al tanto de la posibilidad de una deficiencia de ácidos grasos esenciales secundaria a una dieta libre de grasas (7).
Por ello, Wolman Moshe recomienda la aplicación percutánea diaria de aceites ricos en ácidos grasos insaturados como aceite de girasol, alazor, o preferiblemente soya, canola, lino, hígado de bacalao o alga, en la piel de una extremidad diferente cada día iniciando con un pequeña dosis (10-15 ul) la cual puede ser duplicada cada cierto número de días, y señala además que el calentamiento de la piel luego de su aplicación contribuye a su absorción. No está claro si una adherencia estricta a este régimen terapeútico es mandatoria, pero es posible que los triglicéridos absorbidos sean metabolizados por la epidermis y transportados en ciertas cantidades a otros órganos en una forma metabolizable. Por otra parte, se ha mencionado la posibilidad de utilizar un inhibidor de la HMG-CoA reductasa como la lovastatina, a dosis de 20 mg BID VO como tratamiento coadyuvante. El mecanismo de acción se basa en la disminución de la síntesis de colesterol y de la producción de la apolipoproteína B con reducciones en los niveles tanto de colesterol como de triglicéridos. Asimismo, se ha evidenciado una disminución de la heptoesplenomegalia y una mejoría de la disfunción adrenal (14,18).
Algunos autores citan que la enfermedad de Wolman es también una de las candidatas para terapia genética dirigida al hígado. En un estudio realizado en el 2001 por la Universidad de Pensylvania se probaron los efectos de terapia génetica de reemplazo en fibroblastos deficientes de lipasa ácida lisosomal (LAL) utilizando como vector un adenovirus recombinante que codifica al cDNA humano de la LAL. Se evidenció un aumento dosis-dependiente en la actividad hepática de la LAL y 72 horas posterior a su inyección se demostó corrección del acúmulo de lípidos en los fibroblastos, así como disminución de la hepatomegalia y normalización de la histopatología (19,9,22).
Asimismo, en otro estudio similar llevado a cabo en "The Children's Hospital Research Foundation" en Cincinati en el año 2002 se obtuvo una disminución de un 36% del peso hepático, una reducción en el acúmulo de lípidos en los macrófagos que se evidenció en el exámen histológico de los tejidos así como una disminución del 50% en los niveles de triglicéridos y colesterol tanto en el hígado como en el intestino delgado y de un 60% en el bazo (19,20,21).
Se ha ensayado como tratamiento el transplante hepático y de médula ósea sin buenos resultados, aspecto que en parte pudiera ser debido a la toxicidad de la quimioterapia usada en los protocolos de preparación (1). Aunque aún no se dispone de un tratamiento eficaz, es interesante su conocimiento para establecer un adecuado consejo genético, e incluso la posibilidad de un estudio antenatal mediante amniocentesis y análisis enzimático (3,11,13).
Referencias
1. Parada J, Blanco M, Reparaz R et al.. Enfermedad de Wolman: A propósito de un caso. An Esp Pediatr 1996;45:428-30. [ Links ]
2. Valerdiz S, Velasco R y Rodríguez R. Enfermedad de Wolman. Depósito pulmonar de lípidos. An Esp Pediatría 1997; 47: 427-8. [ Links ]
3. Vargas F, Gómez A, Cuevas J y Young EP. Enfermedad de Wolman. An Esp Pediatría 1987; 27: 195-8. [ Links ]
4. Brent Harrison, R. and Francke P. Radiographic Findings in Wolman's Disease. Radiology 1977; 124: 188. [ Links ]
5. Wolman Moshe, M.O. Proposed treatment for Infants with Wolman Disease. Pediatrics 1989; 83: 1074. [ Links ]
6. Dutton R.V. Wolman's disease: Ultrasound and CT diagnosis. Pediatr Radial 1985; 15: 144-146. [ Links ]
7. Wolman Moshe, M.D. Wolman Disease and its treatment. Clin Pediatr 1995; 207-12. [ Links ]
8. Kikuchi M, Igarashi K, Nora T, et al. Evaluation of Jejunal Function in Wolman's disease. J Pediatr Gastroenterol Nutr. 1991; 12: 65-9. [ Links ]
9. Roytta M, Fagerlund AS, Toikkanen S, et al. Wolman disease: morphological, clinical and genetic studies on the first Scandinavian cases. Clin Genet 1992;42: 1-7. [ Links ]
10. Coates P, Cortner J, Mennuti M et Wheeler James. Prenatal Diagnosis of Wolman Disease. Am J Med Genetics 1978;2:397-407. [ Links ]
11. Desai P, Astrin K, Thung S et al. Cholesteryl Ester Storage Disease: Pathologic Changes in a Affected Fetus. Am J Med Genet 1987; 26: 689-98. [ Links ]
12. Hoeg J, Demosky S, Pescovitz O et Brewer B. Cholesteryl Ester Storage Disease and Wolman Disease: Phenotypic Variants of Lysosomal Acid Cholesteryl Ester Hydrolase Deficiency. Am J Hum Genet 1984; 36:1190-1203 [ Links ]
13. Assmann Gerd et Fredrickson Donald. Acid Lipase Deficiency: Wolman's Disease and Cholesteryl Ester Storage Disease. The Metabolic Basis of inherited disease. McGraw Hill Book Company, 1983: 803-819. [ Links ]
14. Pediatric Database. Wolman Disease en: www.icondata.com/health/pedbase [ Links ]
15. Suchy J. Frederick. Liver Disease in Children. Mosby-Year Book Inc. 827-8. [ Links ]
16. CHORUS. Adrenal Calcification en: chorus. rad. mcw. edu/doc/00009. html [ Links ]
17. Beaudet A, Ferry G, Nichols B et Rosenberg H. Cholesterol ester storage disease: clinical, biochemical, and pathological studies. J Pediatr 1977; 90: 910-14. [ Links ]
18. National Library of Medicine: Characterization of Iysosomal acid lipase mutations in the signal peptide and mature polypeptide region causing Wolman disease en: www.ncbi.nlmn.nih.gov [ Links ]
19. Du H. Enzyme therapy for Iysosomal acid lipase deficiency in the mouse. Hum Gene Ther 2002; 10: 1639-48. [ Links ]
20. Du H. Lysosomal acid lipase deficiency: correction of lipid storage by adenovirus mediated gene transfer in mice. Hum Gene Ther 2002; 13: 1361-72. [ Links ]
21. Aslanidis, C. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual Iysosomal acid lipase activity. Genomics 1996; 33: 85-93. [ Links ]
22. Anderson R A, et al. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Molecular Genetics Metabolism. 1999; 68: 333-345. [ Links ]
23. Tiejtge UJ, et al. Phenotypic correction of lipid storage and growth arrest in Wolman disease fibroblasts by gene transfer of Iysosomal acid lipase. Hum Gene Ther 2001 ; 12: 279-89. [ Links ]
24. Essa, Al et al. Wolman's disease: The King Faisal Specialist Hospital and Research Centre Experience" Saudi Arabia, 1998 [ Links ]
25. Krivit W, qt al. "Wolman disease successfully treated by bone marrow transplantation". Minneapolis. Bone marrow transplant. 2000; 26: 567 –570 [ Links ]
26. Yoshida, H. "Genetic lipid storage disease with Iysosomal acid lipase deficiency in rats". Lab. Animal Sci 1990" 45: 664-668. [ Links ]
27. Fauci S. Anthony et al. Harrison: Principios de Medicina Interna. McGraw-Hill-lnteramericana de España, SAU. 2467-75. [ Links ]
