











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkLas hemoglobinopatías son comunes en el trópico, siendo particularmente frecuente los portadores heterocigotos de Hemoglobina S (HbS) y las alfa talasemias. Las alfa talasemias son enfermedades con patrón hereditario autosómico recesivo, caracterizadas por cursar con anemias microcíticas hipocrómicas de variable intensidad.1,2
La hemoglobina New York (HbNY) o hemoglo- bina Kaohsiung es una variante de hemoglobina que ocurre por la mutación en el aminoácido 114 en la cadena de Beta globina, modificando una valina por un ácido glutámico, y fue Identificada por primera vez en 1967 en una familia de ascendencia China.3
Los individuos heterocigotos para hemoglobina New York son clínicamente normales sin alteración hematológica.4,5 Sin embargo, se han descrito complicaciones severas en doble heterocigotos para HbNY y Hemoglobina S (HbS), la cual se ha descrito como una forma grave de enfermedad drepanocítica y se encuentra asociado a complicaciones como retinopatía, e infarto de bazo.6
Si se hereda la hemoglobina New York junto con una deleción de 3 genes alfa, podría provocar una enfermedad severa por Hb H. Para su correcto diagnóstico, es importante contar con al menos dos metodologías diferentes para la detección de variantes de hemoglobina, ya que si se utiliza un solo método como la cromatografía líquida a alta presión (HPLC por sus siglas en inglés) la variante Hb NY podría pasar desapercibida,7 ya que corre en la misma posición que la hemoglobina A normal. En este caso particular, al ser la muestra procesada por el método de electroforesis capilar, se pudo detectar claramente un pico electroforético en la posición donde normalmente corre la Hb NY.
En Costa Rica, aun cuando ya se han descrito anteriormente pacientes con Hb NY;8,9 la alta prevalencia de portadores para HbS y Talasemia hacen que el hallazgo de un doble heterocigoto de hemoglobina NY/ -3,7 Alfa Talasemia, en el Área de Salud de Aserrí sea relevante y amerita seguimiento, con el fin de brindarle al paciente una correcta interpretación de sus índices hematimétricos y un asesoramiento genético oportuno.
Presentación del caso
Mujer de 40 años de edad, de etnia blanca, con ascendientes guanacastecos, asintomática, sin historial familiar de anemia ni de enfermedades crónicas, la cuál se presenta al Laboratorio Clínico de Aserrí para un control de rutina por seguimiento de anemia. Los índices de serie roja del hemograma tales como la hemoglobina (Hb), el Volúmen Corpuscular Medio (VCM) y la Hemoglobina Corpuscular Media (HCM) no son los esperables para el cómputo de Glóbulos Rojos obtenido, lo cual es compatible con una talasemia (Cuadro 1). Los cómputos de leucocitos y plaquetas se encuentran dentro del rango de referencia. Al revisar el histórico del expediente médico del paciente, se evidencia que los índices hematimétricos hacen incurrir al médico tratante a dar seguimiento por anemia ferropriva. Sin embargo, al utlizar el algoritmo diagnóstico del Laboratorio Clínico, dichos valores en conjunto con la morfología de glóbulos rojos (Figura 1b), hacen sospechar de un síndrome talasémico por lo que se refiere una muestra de sangre periférica al Laboratorio de Estudios Especializados e Investigación (LEEI) del Hospital Nacional de Niños (HNN) Dr. Carlos Sáenz Herrera, para su estudio mediante una electroforesis de hemoglobina.
Cuadro 1. Parámetros hematológicos y genotípicos del propositus
| Parámetro | Resultado | Rango de referencia |
|---|---|---|
| Glóbulos rojos (x 106/uL) | 6.4 | 4.0 - 5.5 x 106/uL |
| Hb (g/dL) | 12.9 | > 12 g/dL |
| Hematocrito (%) | 40.3 | 37-47 % |
| HCM (pg) | 20.0 | > 28 |
| VCM (fL) | 63.0 | 80 - 98 fL |
| CHCM (g/dL) | 32.0 | < 37 g/dL |
| RDW-cv (%) | 20.0 | < 16% |
| Hb A (%) | 59.6 | 95 - 98% |
| Hb A2 (%) | 3.2 | < 3.5% |
| Hb F (%) | 0 | < 2% |
| Hb NY (%) | 37.2 | 0 % |
| α Genotipo | α -3.7 α/ α α | α α/ α α |
| β genotipo | β New York / βA | βA / βA |
Valores obtenidos para los índices de serie roja: Hemoglobina (Hb), Hemoglobina Corpuscular Media (HCM), Volúmen Corpuscular Medio (VCM). Los valores obtenidos en la electroforesis de hemoglobina (HbA, HbA2 y Hb F) y Hb New York (Hb NY). El análsis molecular del gen de las cadenas de Alfa globina (HBA) confirma la presencia de una talasemia -3.7 Kb; y el análisis del gen de la Beta Globina (HBB) la presencia de Hb New York.
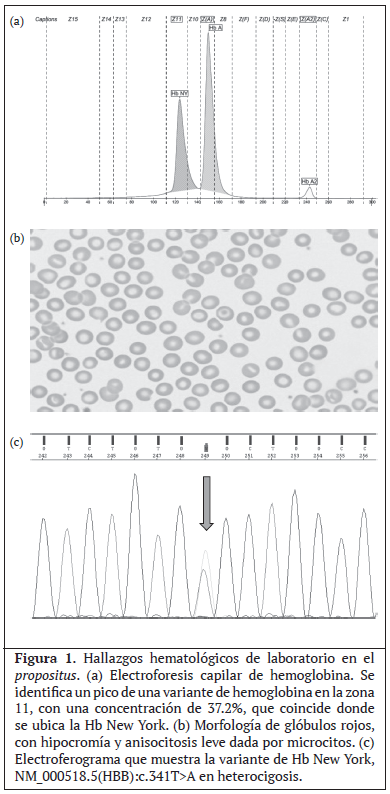
Se realiza electroforesis de hemoglobina por electroforesis capilar (Capillarys 2; Sebia, París, Francia; software versión 6.2). El valor normal obtenido para la HbA2 descarta la presencia de Beta Talasemia, y el valor obtenido para la hemoglobina fetal (HbF) descarta la presencia de una Delta Beta talasemia. Pero la presencia de un pico anormal de hemoglobina en concentración de 37.2% en la zona 11, suguiere la presencia de una variante de hemoglobina conocida como Hemoglobina New York (Figura 1a). Es de notar que la concentración de la HbNY es inferior a lo que se observa en pacientes heterocigotos, pero en concentraciones similares a las que se reportan en casos de dobles heterocigotos HbNY y Alfa Tal -3.7 (7).
Para el análisis molecular por Alfa Talasemia, cuya metodología consiste en la identificación de 21 de las mutaciones / deleciones más frecuentes del gen α-globina basado en amplificación por PC Rehibridación inversa (ViennaLab labor-diagnostikaGmbH, Vienna, Austria); se extrajo ADN genómico de los leucocitos de sangre periférica del propositus y sus progenitores. Mediante PCR e hibridación inversa se identificó la mutación -3,7 heterocigota.
El Laboratorio Nacional de Tamizaje Neonatal y Alto Riesgo realizó la amplificación de las regiones codificantes con cebadores específicos para el gen HBB y posteriormente secuenció mediante electroforesis capilar (Secuenciación de Sanger, AB3500 de Thermo Fisher Scientific) empleando los reactivos BigDye® Terminator v3.1 (Thermo Fisher Scientific). El análisis de secuenciación detectó la variante NM_000518.5(HBB):c.341T>A, la cual cambia el sentido del aminoácido, donde la valina en posición 114 es sustituida por ácido glutámico (GTG>GAG, p.(Val114Glu)); y se considera patogénica causante del fenotipo de hemoglobina New York o Kaohsiung. (Figura 1c).
Discusión:
La Hb New York es muy común en el sur de China,8 y es la variante de hemoglobina más frecuente después de la Hb E, seguida de la Hb Q Tailandia en tercer lugar, por lo que su detección en Asia es común, no así en nuestro país.9 Los genotipos heterocigotos para hemoglobina New York son asintomáticos sin alteración hematológica,9 pero en asociación con otras hemoglobinopatías como alfa o beta talasemias o la hemoglobina S, el individuo manifiesta cuadros más severos.6 La identificación de estas variantes representan un hallazgo importante que permite darle al clínico una interpretación correcta de los índices hematimétricos,para ofrecerle al paciente un adecuado seguimiento clínico, consejería genética y que conozca las posibles implicaciones de su descendencia.
En el caso de nuestra paciente, no se registra en su historial familiar anemia ni enfermedades crónicas. La paciente estaba recibiendo seguimiento por anemia ferropriva la cual no respondía al tratamiento.
De aquí la importancia de aplicar un algoritmo diagnóstico que permita la correcta interpretación de los todos los índices hematimétricos y morfología de glóbulos rojos que permitió identificar una doble heterocigosis por alfa talasemia y Hb NY como causa de anemia microcitica la cual no respondía al tratamiento.
En nuestro país son muy pocos los casos reportados de heterocigotos para Hb NY, y en el análsis de publicaciones nacionales no encontramos registros de ningún caso previo de doble heterocitogo para Hb NY y Alfa Talasemia -3.7 kd, siendo este el primer caso descrito en la literatura científica costarricense. Por las implicaciones clínicas antes descritas, el consejo genético resulta prioritario, y así se le hizo ver a la paciente.
Agradecimiento: a la asistente diplomado Keren Vanegas García, del Laboratorio Clínico del Área de Salud de Aserrí por sus aportes en el procesamiento técnico de la muestra.

