











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkLas cardiopatías congénitas (CC) son la primera causa de defectos congénitos en el mundo, con una prevalencia estimada entre 8 y 9 por 1000 nacimientos, ocupando los primeros puestos como causa de mortalidad infantil.1-6
Avances en las técnicas de diagnóstico, las intervenciones quirúrgicas y médicas y el cuidado neonatal, han incrementado la supervivencia de los niños que nacen con CC alrededor del mundo.7 En los países desarrollados, las CC son más frecuentes en adolescentes y adultos que en niños, con una relación en el año 2005, del 60 % en adultos y el 40 % en niños.8 Reportes indican que el 96 % de los recién nacidos con CC que sobreviven el primer año de vida, alcanzarán los 16 años de vida.9,10
En países en vías de desarrollo, son muy escasos los estudios de mortalidad y supervivencia de CC, pese a que en estos existe una mayor proporción de población infantil, así como mayor prevalencia de CC severas.11
En Costa Rica, un país pequeño de ingreso mediano-alto, con un promedio anual de 72495 nacimientos en la última década12 (98 % intrahospitalarios) y una tasa de mortalidad infantil de 7,76 por cada 1000 nacidos vivos* para el año 2015, las CC son el grupo de defectos congénitos más frecuentes, con una prevalencia de 6 x 1000 nacimientos (IC95 % 5-7 x 1000 nacimientos), 13 que representan cerca del 13 % de la mortalidad infantil.14
Pese a que la cobertura de atención prenatal temprana oscila entre el 68 y el 83% (Trejos ME. Análisis de mortalidad infantil, situación de la salud reproductiva y la planificación familiar. En: Foro de mortalidad infantil, 9 diciembre, 2010; San José, Costa Rica), con un promedio de 6 consultas prenatales, este control no incluye ecografía cardiaca fetal, y el diagnóstico prenatal de la CC es menor al 5 % de los casos.13
El estudio pretende analizar la supervivencia a los cinco años de edad para los niños que nacieron con CC en Costa Rica, entre 2006 y 2007, con el fin de aportar insumos para abordar este creciente problema de salud pública en el país.
Metodología
Se realizó un estudio observacional de cohorte retrospectiva y de base poblacional, para determinar la supervivencia al año y cinco años de edad, de niños nacidos con CC entre el 1 de enero de 2006 y el 30 de junio de 2007 en Costa Rica, diagnosticados antes de un año mediante ecografía cardiaca realizada por un cardiólogo en el Hospital Nacional de Niños (HNN). Para el análisis de supervivencia se tomó como tiempo 0 la fecha de nacimiento y se siguieron los casos hasta agosto de 2012, fecha cuando todos habrían cumplido al menos 5 años de edad (rango de seguimiento de 5 a 6,66 años).
Como fuente de información se utilizó la base de datos de egresos hospitalarios y expedientes médicos del HNN y se cotejó con los datos del Instituto Nacional de Estadística y Censos, centro nacional que registra todas las muertes del país y sus causas, para determinar el estatus de vivo o muerto.
Dado que el HNN es el único centro de referencia para el diagnóstico y tratamiento de los niños con CC en el país, los resultados del estudio tienen inferencia nacional. Para realizarlo se obtuvo aprobación del Comité Ético Científico del Instituto Costarricense de Enseñanza e Investigación en Nutrición y Salud (INCIENSA) y del HNN.
Clasificación de los defectos cardiacos congénitos y casos:
Todas las anomalías fueron codificadas de acuerdo con la Clasificación Internacional de las Enfermedades, décima edición, tomando en cuenta los códigos entre Q200 y Q264. Se excluyeron cardiopatías relacionadas con la posición cardiaca (Q240-Q241-Q248), del ritmo (Q246), de la vena cava (Q260-261), así como miocardiopatías, cardiomegalia y CC inespecíficas (Q248-Q249).
Las CC incluidas se clasificaron como muestra el Cuadro 1.
Los casos se clasificaron además según sexo, edad al diagnóstico (0-6 días, 7-29 días, 30-364 días y 365 o más), grupo de edad de la madre (13-19 años, 20-34 años y 35 o más años), índice de desarrollo social del cantón (municipio) de procedencia materna y estatus quirúrgico como sigue: sin cirugía, cirugía cardiaca cerrada (sin corazón abierto ni circulación extracorpórea) y cirugía cardiaca abierta (con circulación extracorpórea o a corazón abierto).
Análisis estadístico:
Se calculó la prevalencia y mortalidad de CC en la cohorte de nacimiento estudiada con su respectivos IC95 %.
Se determinaron tasas de supervivencia acumulada al primer año y a los cinco años de edad, y se compararon con las de la población general del país, la cual se estimó de las tablas de vida publicadas por el Centro Centroamericano de Población.* Se utilizó el método Kaplan-Meier, se estimaron probabilidades no paramétricas de supervivencia con su respectivo IC95 % a las edades de una semana, cuatro semanas, un año y cinco años. Para cada categoría de diagnóstico específico se tomó en cuenta los casos en donde el defecto no estuviera asociado a otra CC, excepto a la persistencia del conducto arterioso.
Se compararon las funciones de supervivencia entre los diferentes estratos de los factores o covariables. Para evaluar la significancia estadística (p<=0,05) se utilizó la prueba de Log Rank.
Por último, para evaluar potenciales factores asociados a la supervivencia, se utilizó un modelo multivariado de riesgos proporcionales de Cox, utilizando el método de eliminación en reversa (partiendo de un modelo lleno), hasta lograr la mayor significancia del modelo de acuerdo con el valor de -2log de la verosimilitud y el X2 del modelo. Las variables incluidas en el modelo fueron aquellas que en el análisis univariado obtuvieron valores de Hazard Ratio (HR) con una significancia de p≤0,2, determinado por medio del test de Wald. El análisis estadístico se hizo mediante el programa SPSS v20.
Resultados
Desde enero de 2006 hasta junio de 2007 nacieron 544 niños con CC entre 105 777 nacimientos, para una prevalencia de 5,14 por 1000 (IC95 %:4,73 -5,60). De ellos, un total de 543 casos fueron seguidos hasta los 5 años de vida, encontrando una mortalidad del 27,9 % (IC95 %:24,21-31,73; n: 152). La mayoría falleció antes del primer año de vida (85,5 %), 34,9 % de ellos durante el período neonatal (17,1 % primera semana). La media de supervivencia para la cohorte fue de 1802,8 días (IC95 %:1717,5-1888,2).
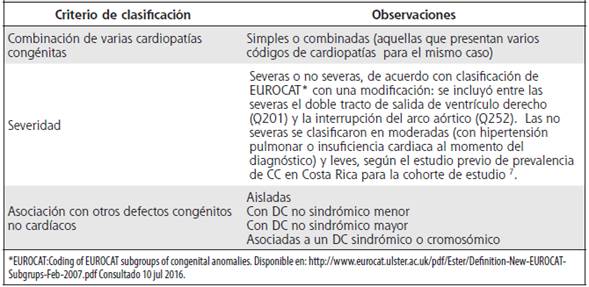
La Figura 1 muestra la función de supervivencia de los niños con CC de la cohorte de nacimientos estudiada. La probabilidad de supervivencia al año de edad fue del 76,1 % versus el 99,1 % población general de un año de vida con y sin cardiopatías para el 2006. Al día 792 (2 años y 6 meses, aproximadamente), la curva de supervivencia se estabiliza en el 72,9 %, alcanzando una supervivencia acumulada del 72,4 %, cuando los niños cumplen 5 años de vida, versus el 98,8 % para la cohorte nacional de nacimientos de 2006 (con y sin CC) de Costa Rica.
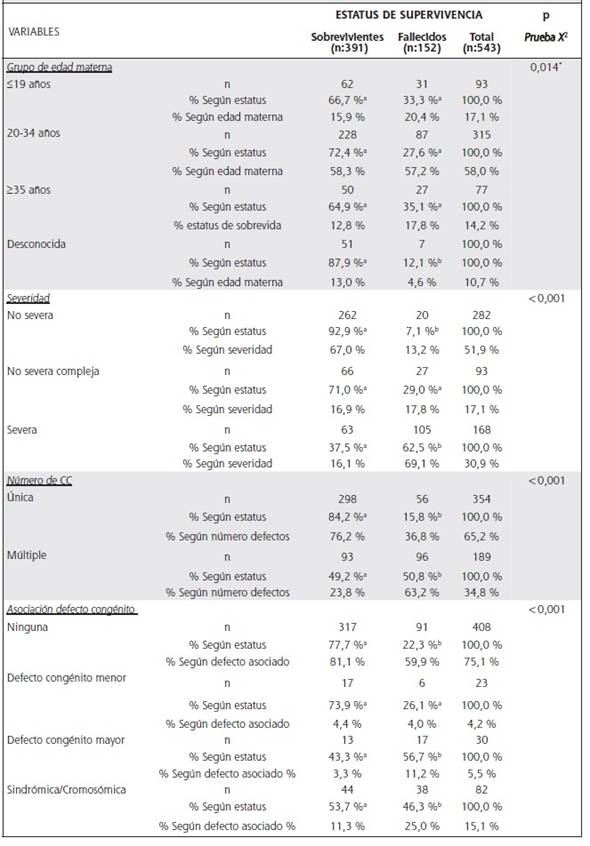
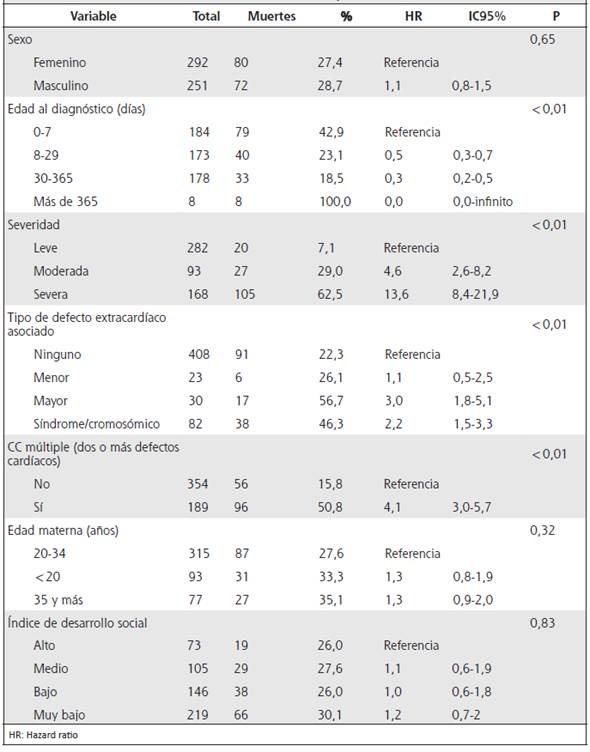
El Cuadro 2 muestra la distribución de la población de estudio en los estratos de las variables de análisis y la frecuencia de censura (supervivencia) entre estratos. Las variables de edad al diagnóstico, severidad de la CC, número de defectos cardiacos del caso (CC múltiple o simple) y asociación de defectos congénitos no cardiacos, presentaron diferencias significativas de supervivencia entre sus estratos; caso contrario sucedió con variables como: sexo, grupo de edad de la madre, índice de desarrollo social del cantón de procedencia materna y procedencia materna (provincia).
El Cuadro 3 muestra la mortalidad y probabilidad de supervivencia en diferentes edades hasta los cinco años de edad de los diagnósticos seleccionados de CC para el estudio, así como para el total de las CC. Analizando la tabla según diagnóstico, la supervivencia más reducida la presentan los niños con transposición de grandes arterias, seguida del canal aurículo-ventricular y la tetralogía de Fallot. Este cuadro ofrece una idea del período de edad cuando el niño es más susceptible de fallecer, según el diagnóstico específico de CC. Por ejemplo, los pacientes con comunicación interventricular (diagnóstico más frecuente encontrado en la cohorte de nacimientos), tienen su período de mayor susceptibilidad entre el primer mes y el primer año de vida, mientras que los pacientes con coartación de aorta lo tienen entre la primera semana y el primer mes de vida.
La Figura 2 muestra la curva de supervivencia de acuerdo con la severidad de las CC. Como era de esperar, hay diferencias altamente significativas entre los tres niveles de severidad, siendo la mayor supervivencia para las CC no severas, seguidas de las CC moderadas y por último, las severas.
La Figura 3 muestra la función de supervivencia de supervivencia de acuerdo a la presencia o ausencia de defectos congénitos extracardiacos, asociados a la CC. Se observa que no hay diferencia en la supervivencia de pacientes con CC sin defectos congénitos asociados y con un defecto congénito menor. Los pacientes que presentan la menor supervivencia son los que asocian defectos congénitos mayores, seguidos de los que asocian defectos congénitos genéticos o cromosómicos.
La mortalidad fue mayor en los niños diagnosticados durante la primera semana (42,93 %) en comparación con los diagnosticados entre los 7 y 29 días (23,12 %), y del mes al año de vida (18,54 %). La mortalidad de los niños diagnosticados después del año de vida fue de cero. La frecuencia de diagnóstico prenatal encontrado en la cohorte fue del 0,37 %; se trató de dos casos, uno con comunicación interatrial e interventricular más persistencia del conducto arterioso, y otro con ventrículo único, ambos con defectos congénitos extracardíacos mayores asociados, que fallecieron a los 19 y 20 días, respectivamente, en el HNN.
Fueron sometidos a cirugía cardiaca 193 niños (35 %), de los cuales fallecieron 80 (41,5 %). La media y mediana de supervivencia de los niños que fallecieron después de la cirugía no sobrepasó los seis meses (199 días (DS 316d) y 80 días, respectivamente). En el análisis de Kaplan Meier, según estatus quirúrgico, presentaron una mayor probabilidad de supervivencia aquellos niños que no fueron operados, lo que podría estar en relación con la severidad del defecto; no obstante, la función o riesgo de supervivencia no difiere significativamente de aquellos niños que se sometieron a cirugía cardíaca a corazón abierto y uso de circulación extracorpórea. Se demostró que el peor pronóstico de supervivencia (p≤0,05 según la prueba de Log Rank) fue para aquellos niños que antes del año de vida tuvieron cirugía cardíaca cerrada, lo cual podría relacionarse con que estas cirugías a menudo son realizadas de emergencia en condiciones clínicas de riesgo (prematuridad, desnutrición, compromiso respiratorio o sepsis), o bien, para paliar síntomas severos y posteriormente hacer la cirugía correctiva, como es el caso de la cirugía para fístulas de Blalock-Taussig-Thomas modificada, cerclaje de la pulmonar y ligadura del conducto arterioso. Estas condiciones conllevan un periodo de estancia hospitalaria prolongado y un alto riesgo de que se infecten y luego fallezcan.
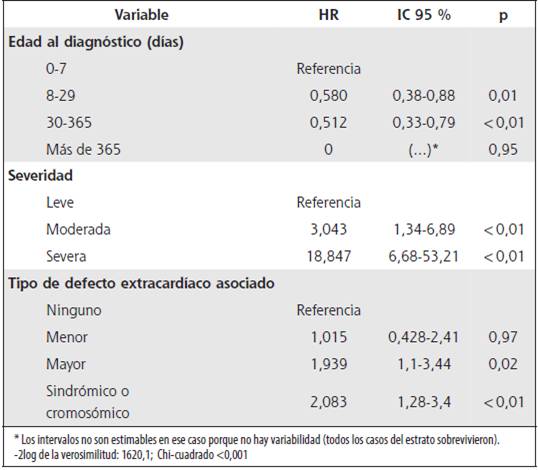
El Cuadro 4 muestra el análisis de riesgo relativo crudo de cada factor estudiado y el Cuadro 5, el riesgo ajustado de acuerdo con el modelo de riesgos proporcionales de Cox, para el cual se partió de un modelo lleno. Los factores significativamente asociados (p <0,01) con una menor probabilidad de supervivencia fueron: la severidad de la CC, la combinación de dos o más CC, la edad temprana al momento del diagnóstico (en ausencia de diagnóstico prenatal o tamizaje neonatal) y la asociación con defectos mayores sindrómicos o cromosómicos.
En el análisis univariado, se exploró la cirugía cardíaca en comparación con la no- cirugía cardíaca (RR = 1,77, IC del 95 % = 1,20-2,60 para cirugía cardíaca a corazón abierto y RR = 2,33, IC del 95 % = 1,58-3,44, para cirugía cardíaca sin corazón abierto). No obstante, en el modelo ajustado por todas las variables descritas, las CC múltiples y la cirugía cardíaca perdieron significancia estadística. No se encontró asociación significativa entre la supervivencia y el sexo, la edad y el origen materno, o el índice de desarrollo social de la ciudad de origen.
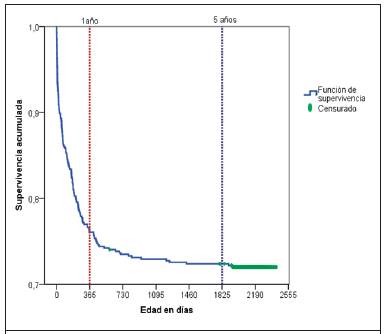
Figura 1 Función de supervivencia al año y cinco años de edad de niños con cardiopatías congénitas , cohorte de nacimieto 2006-2007, Costa Rica.
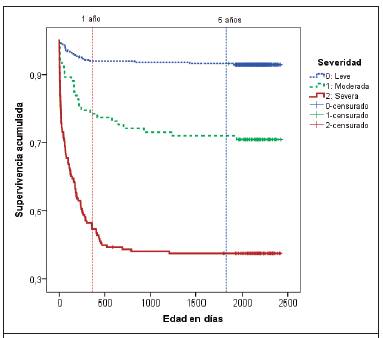
Figura 2 Función de supervivencia de acuerdo a la severidad de la cardiopatía congénita, cohorte de nacimieto 2006-2007, Costa Rica.
Discusión
La prevalencia de CC que arroja el estudio se encuentra dentro del rango reportado a nivel global, el cual se estima entre 7 y 9 por 1000 nacimientos, con una variación entre 4 y 50 casos por 10000 nacimientos, 1-6 de acuerdo con la capacidad de diagnóstico de los casos, si existe o no diagnóstico prenatal, los criterios de inclusión y exclusión de cada registro, así como el tiempo de seguimiento de los niños después del nacimiento.
La mortalidad encontrada (28 %), con una supervivencia al año de vida del 76,1 % y del 72,4 % a los cinco años, está también dentro del rango encontrado por estudios previos de países no industrializados y algunos desarrollados. Los datos mundiales en mortalidad de CC deben interpretarse con cautela dependiendo de las condiciones de atención y tratamiento quirúrgico disponibles en cada región que se estudia, así como de la existencia de diagnóstico prenatal y posibilidad de realizar una terminación electiva del embarazo. La bibliografía mundial reporta diferencias notables de mortalidad por CC entre países industrializados y no industrializados, que van del 3 -7 % al 20 %, respectivamente.2 Por ejemplo, la mortalidad reportada en comunidades de Argentina es del 13,79 %,15 mientras que en Cuba fue del 24 % a los tres años de vida; no obstante, en este país en donde todos los casos estudiados tuvieron diagnóstico prenatal, el 82 % de las madres embarazadas optaron por terminar el embarazo. 16 En México, Mendieta et al17 reportaron en un estudio de base hospitalaria, una mortalidad del 18,64 %; sin embargo, un estudio de base poblacional de la cohorte de nacimientos de 2013, publicado por Torres-Cosme et al,18demostró una mortalidad infantil por CC del 24,8 %, donde aproximadamente un tercio de las muertes sucedieron en la primera semana de vida. Datos similares se reportan en China, donde las CC representan el 42 % de la mortalidad infantil, con supervivencia del 77,2 % al primer año de vida. 19
En países desarrollados el panorama es a veces distinto. En Atlanta, EEUU, Oster et al 20 reportaron una mortalidad al año de CC no críticas del 2,9 % y de CC críticas del 17,5 % (supervivencia del 97 % y del 82,5 %, respectivamente); no obstante, en el estudio se excluyeron las cromosomopatías y pacientes con defectos congénitos no cardíacos mayores. En Arkansas, Cleves et al21 reportan una mortalidad al año de vida del 11,8 % y una supervivencia del 88,2 % (excluyendo niños con agenesia renal, anencefalia y Trisomías 13 y 18). Por otro lado, en Canadá, Agha et al22 reportaron una mortalidad del 25 al 29 %, con una supervivencia al año de vida, de alrededor del 71,2 % y el 85,7 % y del 68,1 % y el 84 % a los cinco años, similar a los resultados de Costa Rica. En general, en los Estados Unidos la mortalidad y supervivencia presentan las cifras más favorables de América.
En Europa, Jortveit J et al,23 en Noruega, estudiaron la mortalidad acumulada al primer año de vida de niños nacidos con CC entre 1994 y 2009, la cual fue del 17,4 %; durante el período de estudio la mortalidad por CC severas descendió cerca de un 4 %, debido al aumento de las terminaciones electivas del embarazo. Un estudio multicéntrico en la región norte del Reino Unido, publicado por Tennant et al,24 en donde siguió por 20 años a la cohorte de nacimientos de 1978 a 1986, arrojó una mortalidad del 9,6 % con una supervivencia al año de edad del 92,3 % y del 91,1 % a los cinco años. En España, Pérez-Lescure J et al,25 publicaron un estudio de base poblacional que tomó en cuenta los egresos hospitalarios de 2003-2012, el cual arrojó una mortalidad por CC al primer año de vida, del 4,58 %, en donde el 73,8 % de los niños falleció en la primera semana de vida.
De igual manera que en el resto de los países, en Costa Rica el período de mayor riesgo de fallecer es el período neonatal y para cardiopatías que requieren intervención quirúrgica temprana. Varios estudios demuestran cómo el diagnóstico prenatal mejora los resultados de estos niños, dado que permite planear el momento, tipo y sitio del parto, así como mejorar las condiciones preoperatorias, disminuyendo la frecuencia de intubación preoperatoria, uso de antibióticos y cateterismos cardíacos.26,27 Sin embargo, la bibliografía no es clara con respecto al diagnóstico prenatal y los resultados postoperatorios y mortalidad, por lo que se presentan muchas veces resultados controversiales. En este sentido, la disminución en la mortalidad después de un diagnóstico prenatal puede obedecer a dos cosas: a que las madres opten por una terminación del embarazo ante un diagnóstico prenatal de CC (lo cual no posible en Costa Rica), o a que efectivamente pueda hacerse una intervención oportuna de este niño antes de que su CC lo descompense en las primeras semanas de vida. En un estudio de cohorte retrospectivo en Atlanta, Wright et al 28 demostraron cómo los casos con diagnóstico prenatal tuvieron una mortalidad significativamente mayor al primer año de vida, y explican que los casos captados por este diagnóstico prenatal son más severos, e incluso poseen más probabilidad de tener algún síndrome identificado, bajo peso al nacer y prematuridad, factores que por sí mismos empeoran el pronóstico de supervivencia. En el estudio, solo 3 niños de los 543 fueron diagnosticados prenatalmente, ya que en Costa Rica no existe un protocolo estandarizado de referencia para diagnóstico prenatal de CC, ni se cuenta en todas las regiones con la capacidad instalada de hacer una ecocardiografía fetal.
Además, el estudio demostró que la probabilidad de supervivencia fue menor cuanto más temprano se realizó el diagnóstico postnatal. Esto podría explicarse ya que cerca de la mitad de los niños con CC no presentan síntomas o signos al examen neonatal realizado en las maternidades.29,30 Además, las CC severas o críticas son las que presentan descompensación más temprano después del alta de las maternidades, después de las 12 a 24 horas de vida,31 lo cual conlleva a rehospitalizaciones dentro de las dos primeras semanas de vida, asociadas a un deterioro de su condición clínica, incrementándose la probabilidad de morir, por una intervención poco oportuna. A su vez, estudios han demostrado que entre el 10 y el 30 % de los niños que mueren por CC no son diagnosticados antes de morir o sufrir un compromiso fisiológico serio pre mortem.32 Por esta razón, una de nuestras recomendaciones para mejorar la supervivencia de los niños con CC severas, es implementar el tamizaje neonatal para CC críticas mediante oximetría de pulso, ya que este tamizaje ha demostrado que es una herramienta efectiva para detectar este tipo de CC y optimizar el abordaje de estos niños de manera oportuna.33
Por último, otro factor vinculado a baja supervivencia fue la asociación de defectos cromosómicos o defectos congénitos mayores no cardíacos. Esto implica que el abordaje del niño con CC debe ser integral y conviene descartar oportunamente la asociación de defectos congénitos mayores asociados, así como mejorar el consejo genético para las familias de estos niños.
El carácter observacional y retrospectivo del estudio, basándose en registros médicos, es una limitación a su alcance, ya que se puede haber subregistrado casos de CC leves, o se pueden haber perdido casos de CC severas en niños nacidos y fallecidos en un hospital regional, sin poder tener una confirmación diagnóstica en el HNN. No obstante, según registros del HNN, estos casos no llegan al 5 %.
En conclusión, la supervivencia de los niños nacidos con CC ha venido mejorando por la práctica de estrategias diagnósticas y terapéuticas oportunas, y por avances tecnológicos médicos y quirúrgicos. Sin embargo, persisten inequidades entre países desarrollados y en vías de desarrollo, donde son muy escasos los estudios de supervivencia de niños con CC y sus determinantes.
Los niños costarricenses que nacieron con CC en la cohorte de nacimientos estudiada presentaron una cifra alta de mortalidad, y el peor pronóstico de supervivencia fue en aquellos que necesitaban una cirugía cardíaca a temprana edad. Se deben hacer esfuerzos para implementar un programa de ecocardiografía fetal de calidad en los hospitales regionales, con el fin de mejorar el diagnóstico prenatal, de manera que se consiga planear el sitio, momento y forma de parto. Así mismo, es urgente optimizar la cobertura, calidad y seguimiento de los programas de tamizaje neonatal de CC críticas, e implementar protocolos estandarizados de manejo médicos pre y postquirúrgicos para estos niños.
Cuadro 3 Estimados de supervivencia en diferentes edades hasta los cinco años para niños nacidos con cardiopatías congénitas, cohorte de nacimientos 2006-2007, Costa Rica

Cuadro 4 Análisis univariado en función del tiempo de vida / censura, de factores asociados a la supervivencia a cinco años de edad de niños con cardiopatías congénitas en Costa Rica. Cohorte de nacimientos 2006 a jun 2007
Conflicto de interés: los autores declaran no tener conflicto de interés.

