











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkLos productos naturales son conocidos por jugar un papel muy importante en la medicina tradicional de los diferentes lugares, ya que el conocimiento acerca del uso medicinal que las plantas presentan se ha ido transmitiendo a través del tiempo. Una de estas plantas es la sábila (Aloe vera), la cual es reconocida en el nivel mundial por tener una gran cantidad de cualidades medicinales, entre las que se pueden destacar su actividad antioxidante y antiinflamatoria, además de que protege el sistema digestivo y mejora la cicatrización de las heridas (Joseph y Raj, 2010; Shariff, 2010).
La sábila ha sido utilizada recientemente para la elaboración de productos alimenticios, debido a su acción regenerativa y antioxidante. El estudio fitoquímico de la sábila ha demostrado que la mayoría de sus propiedades medicinales se debe, principalmente, a dos tipos de compuestos: las antraquinonas y los polisacáridos (Femenia, Sánchez, Simal y Roselló, 1999; Hamman, 2008; Ni, Turner, Yates y Tizard, 2004; ITIS, 2017). No obstante, la presencia de una antraquinona, la aloína, un metabolito con acciones no deseadas en los productos de consumo público, hace necesaria la evaluación del producto de una manera más específica. La aloína es un glicósido antraquinónico que al entrar en contacto con las paredes del intestino grueso produce un efecto laxante severo en el organismo (Celestino et al., 2013; van Gorkom, de Vries, Karrenbeld y Kleibeuker, 1999); además, se han descubierto efectos hepáticos (Yang et al., 2010) y carcinógenos de tal compuesto en diferentes estudios, como los realizados por Schörkhuber, Richter, Dutter, Sontag y Marian (1998), Buenz (2008) y el Programa Nacional de Toxicología de los Estados Unidos (NTP, 2001).
Según el Codex Alimentarius, elaborado por la Organización para la Agricultura y la Alimentación (FAO, por sus siglas en inglés) de la Organización de las Naciones Unidas (ONU) y la Organización Mundial de la Salud (OMS), la concentración máxima permitida de aloína en cualquier tipo de alimento (sólido o líquido) es de 0,10 mg/kg (FAO, 1991). La certificación actual otorgada por el International Aloe Science Counsil (IASC) es que la concentración permitida en los productos de consumo oral no debe exceder 10 mg/L de aloína, dato determinado después de obtener información de diversos estudios realizados por la FDA y el Centro Nacional de Investigación Toxicológica de los Estados Unidos, acerca de su toxicidad en ratones (IASC, 2010). Por esta razón, empresas dedicadas a la producción de los suplementos alimenticios basados en el Aloe vera se han dado a la tarea de buscar diferentes metodologías para realizar una cuantificación adecuada de la aloína en los productos que elaboran.
Se han desarrollado múltiples metodologías, utilizando técnicas como cromatografía de gases, cromatografía de capa fina, espectrofotometría y cromatografía líquida. Se pueden mencionar los estudios de Bustamante Botero y Carrascal (2010); Genovese et al. (2010); El Sohly, Gul y Murphy (2004); El Sohly, Gul, Avula y Khan (2007); Ramírez Durón, Ceniceros Almaguer, Cavazos Rocha, Silva Flores y De Torres (2008), y, más recientemente, Mandrioli, Mercolini, Ferranti, Fanali y Raggi (2011), Chiang et al. (2012) y Brown et al. (2014), los cuales han desarrollado y validado métodos para la cuantificación de la aloína en los productos alimenticios.
Costa Rica posee entre 60 y 80 hectáreas de Aloe vera sembradas para su utilización en el nivel industrial, mayoritariamente en suplementos nutricionales. La compañía Natural Aloe, ubicada en Liberia, produce alrededor de 1500 kg por año de extracto de sábila, que son vendidos en los Estados Unidos, principalmente, aunque también se envía a países asiáticos como Corea y China (Natural Aloe, 2017). Actualmente, en el país no hay un laboratorio, público o privado, que tenga implementado un método validado para el análisis de la aloína, por lo que es necesario enviar muestras al exterior para que puedan ser analizadas y se consiga la certificación internacional. Tampoco se han reportado estudios acerca de los productos alimenticios elaborados con sábila (Aloe vera) que se venden en Costa Rica, por lo que se desconoce el contenido de aloína en estos.
Debido a lo anterior, en el presente estudio se lleva a cabo la validación de un procedimiento de análisis cromatográfico (HPLC-DAD) para evaluar la presencia y concentración de aloína en el Aloe vera de productos alimenticios sólidos y líquidos a base de sábila.
Metodología
El método de análisis seleccionado para la validación se basó en el descrito por Metcalfe (2002), con modificaciones implementadas en él. Se utilizó un cromatógrafo líquido Perkin-Elmer Series 200, con una fase móvil de acetonitrilo y una disolución de metabisulfito de sodio al 0,1 % m/v 25:75. Se ensayaron dos columnas: una SupelcoHypersil C-8 (250 mm x 4,6 mm, 5 µm de espesor); otra PhenomenexSpherex C-8 (150 mm x 4,6 mm, 10 µm de espesor). La temperatura de las columnas fue de 35 °C, un flujo de 1,0 mL/min y un detector de arreglo de diodos (DAD). Los ensayos se realizaron a una longitud de onda de 355 nm.
Los estándares para el método se prepararon a partir de una disolución madre de 100 mg/L, utilizando un estándar de aloína 97 %, marca Sigma Aldrich, el cual fue disuelto con una disolución de metabisulfito de sodio 0,1 % m/v. Se prepararon disoluciones patrón con concentraciones entre los 0,1 mg/L y 15 mg/L de aloína.
Selectividad
El análisis de la selectividad se basó en el de blancos, que se le denomina a la mezcla inyectada, la cual contiene todos los reactivos utilizados en el método, pero no el analito de interés. Se realizó el análisis de interferencias en seis blancos de la muestra, un triplicado por cada una de las curvas generadas, asegurando así la selectividad que permite obtener datos menores al límite menor de cuantificación (Vega, 2009).
Linealidad
La linealidad fue determinada por medio de la preparación de curvas de calibración de patrones cuyas concentraciones estuvieron delimitadas por el límite de cuantificación seleccionado, en donde el coeficiente de correlación (R) promedio se utilizó como criterio de linealidad (Vega, 2009). Por un período de 15 días, se realizó una curva por día, inyectándose cada patrón por triplicado. La verificación estadística fue realizada por medio del parámetro estadístico FANOVA (Miller y Miller, 2002; Morales, 2001; Vega, 2009).
Cedasticidad
Para determinar si el comportamiento del equipo utilizado es homocedástico o heterocedástico en los niveles de concentración requeridos, fue utilizada la prueba de Cochran como parámetro de medición.
Límites de detección y límite de cuantificación
Los límites de detección y cuantificación se calcularon con el método estadístico descrito por Meier-Zünd, que establece diferentes relaciones estadísticas, obtenidas a partir de cuatro curvas de calibración de patrones de diferentes concentraciones, los cuales fueron inyectados cinco veces para cada curva (Meier y Zünd, 2005).
Precisión y repetitividad
La precisión del método fue realizada calculando el coeficiente de variación entre los valores obtenidos de los patrones utilizados en el análisis. Se evaluó la repetitividad intradía, inyectando cada uno de los patrones tres veces el mismo día, esto se llevó a cabo para todas las curvas realizadas durante el experimento. Para evaluar la repetitividad interdía, se inyectó los patrones durante tres días consecutivos por triplicado (Ramírez Durón et al., 2008).
Todos los ensayos fueron realizados utilizando las dos diferentes columnas mencionadas anteriormente (SupelcoHypersil C-8 y PhenomenexSpherex C-8), con el fin de comparar los resultados de su utilización y observar diferencias tanto de la aplicabilidad como de la robustez del método.
Se evaluaron dos procedimientos de extracción de aloína, con el objetivo de determinar el más efectivo para las matrices que se trabajaron. Los métodos seleccionados para esta etapa se basaron en los utilizados por El Sohly et al. (2007) y Ramírez Durón et al. (2008).
El procedimiento A de extracción basado en El Sohly et al. (2007) consistió en medir en una balanza analítica una masa entre los 0,25 g y los 0,50 g de muestra sólida y disolverla en 1 mL de agua desionizada. A cada muestra se le adicionó 1 mL de metanol, 2 mL de una disolución saturada de cloruro de sodio y 4 mL de una disolución de acetato de etilo y metanol (9 + 1). Esta mezcla fue agitada en un vórtex durante 30 segundos, luego se dejó en reposo, la fase orgánica se trasvasó a un tubo de ensayo limpio y la fase acuosa fue extraída dos veces más con 2 mL de la mezcla mencionada. Después de combinar las extracciones, se llevó el producto a sequedad en un rotavapor Büchi y el residuo fue disuelto en un baño ultrasónico Branson, utilizando 0,3 mL de metanol y filtrado con un algodón, para luego ser inyectado en el HPLC.
Por otro lado, el método de extracción B basado en el de Ramírez Durón et al. (2008) consistió en medir la masa de aproximadamente 1,00 g de la muestra en una balanza analítica, a la cual se le realizaron dos extracciones con una disolución de etanol 90 % v/v a temperatura ambiente por 5 minutos, con agitación constante; posteriormente, se filtró la muestra en papel Whatman # 1 y se concentró en un evaporador Büchi, manteniendo la temperatura del baño a 35 °C. El sólido obtenido se redisolvió con 10 mL de la disolución de etanol al 90 % v/v y se diluyó en una proporción 1:50 con una disolución de metabisulfito de sodio al 0,1 % m/v, para luego inyectarlo en el HPLC.
Se utilizó un estándar interno, agregando 10 μg de aloína a las muestras que no presentaran señales de este metabolito. Tal adición se realizó de dos maneras: a la primera se le agregó el estándar antes de iniciar la extracción de la muestra; a la segunda el patrón le fue adicionado posterior a la extracción. Se utilizaron dos muestras diferentes (una sólida y una líquida) por triplicado y el procedimiento se llevó a cabo en tres días diferentes, calculando el porcentaje de recuperación (% R) en ambos casos.
Posterior a la validación del método analítico seleccionado, se realizó el análisis de la aloína en los diferentes tipos de productos, ya sean productos finales para consumo o materias primas de proceso, elaboradas a base de sábila. Se realizó un muestreo aleatorio, seleccionando 14 productos de consumo humano de distintas casas comerciales, tanto nacionales como internacionales. Se muestrearon envases de 500 mL para las muestras líquidas y para las muestras sólidas se colectaron envases con un mínimo de 5 gramos. Todas las muestras fueron almacenadas en el laboratorio entre 10 ºC y 15 °C, para mantenerlas en estado óptimo. En la tabla 1, se describe la codificación utilizada, tipo de muestra (sólida, líquida o gel) y su país de origen.
Tabla 1 Muestras seleccionadas para la evaluación de la concentración de aloína
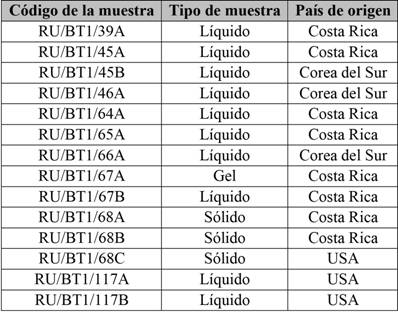
Nota: Fuente propia de la investigación.
Las muestras líquidas fueron analizadas directamente en el HPLC, mientras que las muestras sólidas fueron inicialmente extraídas con el método optimizado y posteriormente analizadas mediante el procedimiento validado para determinar la concentración de aloína. El análisis se realizó por triplicado para cada muestra. El límite máximo permitido de aloína por el IASC es 10 mg/L, tanto para líquidos como para sólidos o geles, por lo que se comparó la concentración obtenida en las muestras con este valor de referencia.
Resultados y discusión
Los resultados de los parámetros evaluados durante la validación del método de análisis de aloína se resumen en la tabla 2.
Tabla 2 Criterios analíticos utilizados para la validación del método de análisis para aloína en productos alimenticios. *Columna SupelcoHypersil
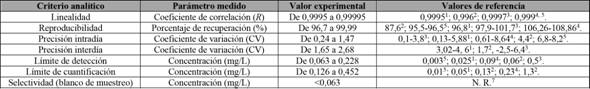
1 El Sohly et al. (2007), 2Ramírez Durón et al. (2008), 3Chiang et al. (2012), 4Brown et al. (2014), 5Mandrioli et al. (2011) , 7no reportado.
Nota: Fuente propia de la investigación.
La selectividad del método se determinó con la inyección de los blancos en diferentes días. En total, se inyectaron 40 blancos por triplicado durante el transcurso del experimento, los cuales no dieron señal alguna con el tiempo de retención del analito. No se encontraron datos reportados en las referencias consultadas para este parámetro. También, se realizó la comparación con muestras que no tuvieran aloína en una concentración mayor al límite de detección determinado. Con el propósito de observar esto, se agregó un estándar interno de 1,00 ± 0,01 mg/L de aloína, para determinar que no se presentan interferencias en el momento de realizar el análisis.
En la figura 1, se observa dos cromatogramas de la muestra RU/BT/45A, entre los cuales hay uno de la muestra sin estándar interno (A) y otro con la adición del estándar interno (B). La selectividad del método hace que el analito sea posible de identificar, ya que se separa completamente de los demás compuestos presentes en la matriz.
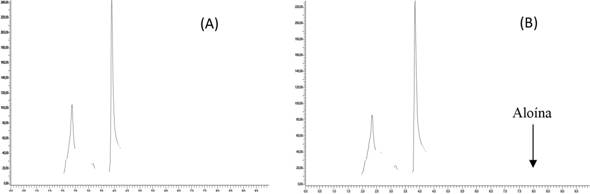
Figura 1 Cromatogramas de la muestra RU/BT1/45A, (A) sin estándar interno, (B) con un estándar interno de aloína de 1,00 ± 0,01 mg/L.
Nota: Fuente propia de la investigación.
Como se observa en la figura 2, en la curva de calibración se obtuvo un coeficiente de correlación mayor que 0,9995, lo cual es indicativo de que el método cumple con las condiciones de linealidad aceptadas en las diferentes fuentes de referencia consultadas, ya que para todas las otras metodologías ensayadas no se presenta un coeficiente menor a 0,9995.
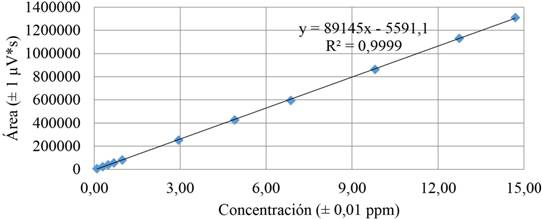
Figura 2 Curva de calibración promedio obtenida para el análisis de aloína, elaborada a partir de una disolución madre de aloína de 98 ± 1 mg/L.
Nota: Fuente propia de la investigación.
El límite de detección para este método es comparable con los rangos que se obtienen en las literaturas consultadas, por lo que la metodología es válida para el análisis cualitativo de muestras, tanto líquidas como sólidas. De la misma manera, el límite de cuantificación para el método utilizado en este análisis está dentro de los parámetros obtenidos en la literatura reportada por El Sohly et al. (2007), lo cual indica que la metodología aplicada es válida para realizar el análisis cuantitativo de aloína en diferentes matrices.
Se utilizó el porcentaje del coeficiente de variación para determinar la repetitividad o la precisión intradía del método (Peters y Maurer, 2002). Al utilizar los patrones de 5,00 ± 0,05 mg/L como muestra de medición, se observó en los resultados que ninguno de los datos excede el 2 % de variación, lo cual sugiere que tal procentaje tiene una excelente precisión, en comparación con lo reportado por El Sohly et al. (2007), Ramírez Durón et al. (2008), Mandrioli et al. (2011), Chiang et al. (2012) y Brown et al. (2014), ya que en varios de los datos reportados se tienen coeficientes de variación mayores, con valores de hasta 5 % y 8 %, en algunos de los casos (tabla 2). Además, se determinó la precisión interdía de la metodología implementada en el ensayo, donde los valores del coeficiente de variación para el método se encuentran entre los más bajos.
Los patrones elaborados durante los diferentes ensayos presentaron resultados con una precisión interdía bastante buena, con coeficientes de variación entre el 1,65 % y el 2,68 %, a pesar de no haber sido elaborados el mismo día de la inyección. Lo anterior indica un buen resultado en este parámetro para las disoluciones que se están utilizando como patrones, por lo cual, con base solo en este aspecto, sería posible utilizar una disolución patrón durante al menos tres días, partiendo del tiempo en que se desarrolló esta parte del ensayo.
La repetitividad del método se determinó con el porcentaje de recuperación. En este caso en particular, se estableció que los porcentajes de recuperación exceden en todos los casos el 96,5 %, indicando un alto nivel de exactitud con los patrones. Los resultados son comparables con los reportados en la tabla 2 por El Sohly et al. (2007), Ramírez Durón et al. (2008), Mandrioli et al. (2011), Chiang et al. (2012) y Brown et al. (2014), en los que se aprecian rangos entre un 86 % y un 108 %, aceptados entre las evaluaciones en el nivel internacional.
El último parámetro analítico evaluado en la calidad del método fue la cedasticidad del sistema. Con el afán de realizar esta medición, se llevó a cabo la prueba de Cochra y se obtuvo un resultado para el equipo analítico de 0,3987, el cual, según los datos consultados, está por debajo del valor crítico aceptado en un nivel de confianza del 95 %, que corresponde a 0,4214, con miras a una cantidad de datos igual a 10. Esto indica que el método en sí es homocedástico y que no hay diferencias significativas entre las varianzas de las concentraciones de los patrones utilizados para evaluar la concentración de aloína.
También, se realizó la adaptación del sistema, utilizando una columna con características diferentes, con el fin de demostrar la robustez de aquel. En la tabla 3, se muestran los parámetros analíticos obtenidos para el método adaptado con la columna PhenomenexSpherex.
Tabla 3 Criterios analíticos utilizados para la validación del método de análisis para aloína en productos alimenticios. *Columna PhenomenexSpherex
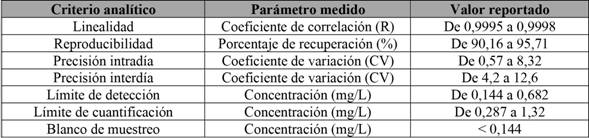
Nota: Fuente propia de la investigación.
Al comparar los resultados de las tablas 2 y 3, se observa, en todos los casos, una concordancia con los valores obtenidos en las diferentes fuentes de literatura consultadas (El Sohly et al. (2007) ; Ramírez Durón et al. (2008); Mandrioli et al. (2011); Chiang et al. (2012); Brown et al. (2014)). Es posible encontrar algunas variaciones en parámetros como el aumento de los límites de detección y cuantificación, los porcentajes de recuperación y los coeficientes de variación en las repeticiones intradía e interdía; sin embargo, realizando una prueba de valor P para estos datos, se determinó que no existen diferencias estadísticamente significativas para ninguna de las pruebas evaluadas.
Se utilizaron dos muestras sólidas para realizar las diferentes extracciones por triplicado: RU/BT1/68A y RU/BT1/68B, utilizando el método validado para la cuantificación de la aloína y, en los casos requeridos, la adición de 500 µL de la disolución madre de 100 ± 1 mg/L de aloína, lo cual daría una concentración final de 5,00 ± 0,05 mg/L como estándar interno.
Los datos de concentración obtenidos para los extractos elaboados son los que se presentan en la tabla 4. Es posible observar que, en el método B, sí se obtienen señales de aloína para la muestra RU/BT1/68B, aunque estas fueron bajas, por debajo del límite de cuantificación. Mientras, para la muestra RU/BT1/68A, se obtuvo un valor promedio de 0,8 ± 0,1 mg/L de aloína. En ambos casos, los datos se encuentran entre los parámetros reportados para este tipo de muestras, según información de Brown et al. (2014), que obtuvo resultados entre 0,00 mg/L y 6,68 mg/L.
Tabla 4 Concentración de aloína para las muestras RU/BT1/68A y RU/BT1/68B, tratadas con diferentes metodologías de extracción
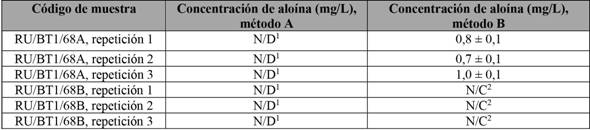
1No detectable, 2no cuantificable.
Nota: Fuente propia de la investigación.
Tampoco se detectaron señales cuando se realizó la extracción por el método A, indicando que esta metodología no es la más idónea por utilizar con las muestras que se tienen, ya que la eficiencia de la extracción es baja. Lo anterior se confirma con los resultados de la tabla 5, para las muestras inyectadas con un estándar interno, con el fin de medir el porcentaje de recuperación de aloína. Se observa que la extracción de aloína en este tipo de matrices es más eficiente con el método B.
Tabla 5 Concentración de aloína con la adición de un estándar interno de (5,00 ± 0,05) mg/L de aloína y su respectivo porcentaje de recuperación
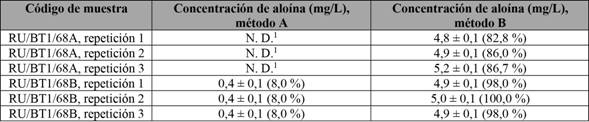
1No detectable.
Nota: Fuente propia de la investigación.
Los productos fueron procesados y analizados según el protocolo implementado de extracción y análisis. Los resultados para las muestras se observan en la tabla 6. Dos productos (RU/BT1/39A y RU/BT1/45B) presentaron valores mayores a los 10 mg/L, límite máximo de concentración de aloína permitido por el International Aloe Science Counsil (IASC, 2010). Una de las muestras no presentó concentración cuantificable (RU/BT1/68A) y otra reporta una no detectable (RU/BT1/68C).
Tabla 6 Determinación de la concentración de aloína (mg/L) por HPLC en productos alimenticios
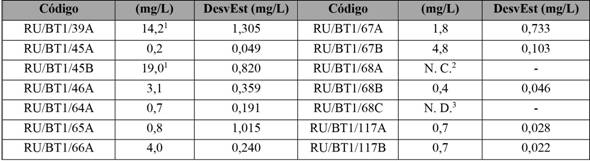
1Valores por encima del mínimo aceptado por el IASC (˂ 10 mg/L). 2No cuantificable. 3 No detectable.
Nota: Fuente propia de la investigación.
En algunos casos, fue posible encontrar productos en los cuales el lavado del gel y el tratamiento con carbón activado era lo bastante eficaz para eliminar el látex que contiene la mayor proporción de aloína (Quesada Espinoza, 2006). En el nivel internacional, las productoras de las bebidas deben cumplir con los diversos estándares para exportar, pero, en cuanto a aloína se refiere, Costa Rica no realiza un análisis de concentración para productos de consumo humano, ya que no hay un laboratorio de control de calidad que certifique los contenidos del metabolito. Debido a lo anterior, no es posible, por el momento, determinar que los productos comercializados se encuentren dentro de los estándares internacionales para la seguridad del consumidor. La presente investigación podría ser una herramienta metodológica para implementar de los controles fitosanitarios respecto al contenido de aloína en alimentos de consumo humano.
Conclusiones
Se implementó satisfactoriamente un método de extracción y análisis de la aloína en productos alimenticios por HPLC, tanto para sólidos como para líquidos elaborados a partir de sábila. La evaluación de la calidad analítica del método demostró valores aceptables, tomando como referencia los reportados por otros autores. El cambio de columnas en el análisis no varía la estabilidad del método, lo que aumenta el nivel de confianza que se puede tener en los resultados obtenidos, ya que se comprobó la estabilidad de los datos.
Resulta importante la implementación de métodos de control, tanto en el nivel gubernamental como en el privado, la cual permita evaluar la calidad de los productos, de una manera más específica, para lograr tener un control de aquellos que se ofrecen en el mercado costarricense.

