











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Los numerosos estudios sobre especies medicinales aún son pocos en comparación con todo lo que falta por conocer acerca de los procesos de síntesis de metabolitos secundarios, tanto en términos de las rutas metabólicas y enzimas involucradas como de los factores internos y externos que afectan su acumulación (Crispin & Wurtele, 2013). En consecuencia, recientemente se ha adicionado la secuenciación y análisis de transcriptomas, ya sea por mapeo contra un genoma o bien mediante el ensamblaje de novo, como una estrategia innovadora para generar información sobre la expresión de genes de ruta como de regulación (Sitakanta et al., 2006).
En Stevia rebaudiana se han identificado más de 100 compuestos, siendo los más conocidos los esteviol glucósidos, que comprenden el esteviósido, los rebaudiosidos A, B, C, D, E y F y dulcósido A, compuestos de sabor dulce, cuyas concentraciones varían ampliamente dependiendo del genotipo y el medio ambiente de producción de la planta (Karaköse et al., 2011; Puri et al., 2011; Wölwer, 2012).
En la ruta metabólica de síntesis de glicósidos en esta especie participan numerosos genes y enzimas, algunos de los cuales han podido ser identificados y caracterizados. Entre estos destacan los denominados SrDXS, SrDXR, SrCPPS, SrKS, SrKO y las tres glucosiltransferasas (UGTs) SrUGT85C2, SrUGT74G1 y SrUGT76G1; además, recientemente se dio la clonación de otros siete genes, SrMCT, SrCMK, SrMDS, SrHDS, SrHDR, SrIDI y SrGGDPS. Para los 15 genes anteriores se ha realizado el respectivo análisis de su expresión dentro de la ruta de esteviol glicósidos (Kumar et al., 2012). Una mejor comprensión de la regulación genética de las vías de biosíntesis podría ser muy útil para manipular el rendimiento de compuestos edulcorantes en Stevia (Guleria & Kumar, 2011; Urban et al., 2013).
Muchos de los compuestos bioactivos de las plantas medicinales se han descrito con detalle gracias a las herramientas de la biología molecular y la bioinformática, que facilitan la identificación de los genes implicados en el metabolismo de compuestos, ya sea de los que participan en la conversión de un compuesto a otro (genes de biosíntesis) o de aquellos que participan en la regulación de la ruta metabólica (genes de regulación) (Muir et al., 2001).
La naturaleza modular de los factores de transcripción ha llevado a la idea de que se puede ejercer un control en la expresión de estos a través de la ingeniería de proteínas, para regular a su vez la expresión del gen o genes de ruta deseados. De manera que coordinar el control de la transcripción de genes de biosíntesis ha surgido como un enfoque eficaz para la producción de metabolitos secundarios en plantas (Sitakanta et al., 2006).
El objetivo de esta investigación fue la búsqueda de posibles mecanismos de regulación de los factores de transcripción, con el fin de ejercer un control que a la vez pueda incidir en el incremento de la expresión de los genes de síntesis de glicósidos.
Materiales y Métodos
El presente trabajo se realizó en el Centro de Investigación en Biotecnología (CIB) del Tecnológico de Costa Rica (TEC) en Cartago, Costa Rica.
Identificación de secuencias génicas reportadas para S. rebaudiana:
La identificación de las secuencias génicas reportadas para la especie S. rebaudiana se hizo empleando la base de datos del National Center for Biotechnology Information (NCBI). La búsqueda se centró en identificar enzimas participantes en la ruta metabólica de síntesis de glicósidos con o sin regiones promotoras descritas.
Análisis de regiones promotoras, identificación de sitios de unión al ADN y relación con factores de transcripción:
Las secuencias génicas reportadas como regiones promotoras en S. rebaudiana fueron analizadas mediante la herramienta Database of Plant Cis-acting Regulatory DNA Elements (PLACE, 2014, Higo et al., 1999) para predecir sitios de unión de factores de transcripción al ADN, que se ubican en los genes de estevia seleccionados. Posteriormente, se empleó la herramienta Plant Interactome Database (Center for Cancer Systems Biology, Dana-Farber Cancer Institute, Harvard Medical School) creada para la especie Arabidopsis thaliana, para relacionar dichos sitios de unión al ADN, con secuencias codificantes de factores de transcripción de A. thaliana.
Secuenciación y análisis de transcriptoma
Material biológico
El material biológico utilizado en la extracción de ARN total correspondió a clones de plantas jóvenes de S. rebaudiana cuyo origen se encuentra en la comunidad El Millón, en Pococí, Limón, Costa Rica. Para la extracción se utilizaron hojas procedentes del segundo y tercer nudo de plantas seleccionadas al azar, mezcladas en una sola muestra.
Extracción de ARN total.
La extracción de ARN total se hizo a partir de 150 mg de muestra de hoja, siguiendo el protocolo Total RNA Isolation from Plant, del NucleoSpin RNA Plant kit marca Macherey-Nagel®. La muestra de ARN se cuantificó con el equipo NanoDrop Lite® y se verificó la presencia de ARN mediante una electroforesis en gel de agarosa al 1,3% durante 45 min a 80 V. Para la precipitación de la muestra se agregó 0.1 V de acetato de sodio 3M a pH 7-8 y 2 V de etanol absoluto.
Secuenciación del ARN
La secuenciación fue realizada por la empresa Axeq Technologies, con el equipo Illumina HiSeq 2000 Sequencer. El software utilizado fue el Illumina Pipeline (CASAVA) v1.8.2.
Análisis de calidad de la secuenciación
La calidad de los fragmentos de secuenciación se verificó con las herramientas Fastqc (Babraham Institute) y RobiNA (Lohse et al., 2010). Se realizaron cortes tipo clipping de la base 1 a la base 15 empleando la herramienta FastX-Toolkit (Hannon Lab) y cortes tipo trimming eliminando colas con calidad inferior a 30% con RobiNA; el filtrado de fragmentos inferiores a las 70 bases también se hizo con esa herramienta.
Alineamiento de genes contra el transcriptoma
El alineamiento de fragmentos se realizó contra los genes participantes en la ruta metabólica de síntesis de glicósidos usando la herramienta RobiNA.
Resultados
Identificación de secuencias génicas reportadas para S. rebaudiana:
La búsqueda bioinformática de genes de S. rebaudiana determinó que existen al menos 20 genes de la ruta de síntesis de glicósidos descritos para esta especie (cuadro 1). La función asociada a dichos genes se muestra en el cuadro 2.
Cuadro 1 Genes de Stevia rebaudiana reportados en el National Center for Biotechnology Information.
| N° de gen | Nombre del gen | Accesión | Promotor descrito | Accesión del promotor |
|---|---|---|---|---|
| 1 | UDP-glicosiltransferasa 79A2 | AY345985.1 | No | - |
| 2 | UDP- glicosiltransferasa 89B2 | AY345983.1 | No | - |
| 3 | UDP- glicosiltransferasa 88B1 | AY345981.1 | No | - |
| 4 | UDP- glicosiltransferasa 73E1 | AY345979.1 | No | - |
| 5 | UDP- glicosiltransferasa 76H1 | AY345977.1 | No | - |
| 6 | UDP- glicosiltransferasa 85A8 | AY345975.1 | No | - |
| 7 | UDP- glicosiltransferasa 85C1 | AY345984.1 | No | - |
| 8 | UDP- glicosiltransferasa 91D1 | AY345980.1 | No | - |
| 9 | UDP-glicosiltransferasa 71E1 | AY345976.1 | No | - |
| 10 | 4-difosfocitidil-2-C-methil-D-eritritol kinasa | DQ269453.5 | Si | FJ755687.1 |
| 11 | IPP/DMAPP sintasa (ispH) | DQ269451.4 | Si | FJ755690.1 |
| 12 | geranilgeranil difosfato sintasa | DQ432013.3 | Si | FJ755692.1 |
| 13 | copalil pirofosfato sintasa (Cpps1) | AF034545.1 | No | - |
| 14 | kaureno sintasa (KS22-1) | AF097311.1 | No | - |
| 15 | kaureno sintasa (KS1-1) | AF097310.1 | No | - |
| 16 | ácido entkaurenoic 13-hidroxilasa (KA13H) | DQ398871.3 | Si | FJ755693.1 |
| 17 | UDP-glicosiltransferasa 85C2 | AY345978.1 | No | - |
| 18 | UDP-glicosaltransferasa 74G1 | AY345982.1 | No | - |
| 19 | UDP-glicosiltransferasa 76G1 | AY345974.1 | No | - |
| 20 | NADPH citocromo P450 reductasa | DQ269454.4 | Si | FJ755694.1 |
| 21 | Actina | AF548026 | No | - |
Fuente: NCBI (2014).
Análisis de regiones promotoras, elementos en cis y factores de transcripción
De los sitios identificados como posibles sitios de anclaje de factores de regulación de la transcripción, se seleccionaron aquellos descritos en la especie modelo A. thaliana (cuadro 3).
Cuadro 2 Función de 20 genes descritos en el National Center for Biotechnology Information para la especie Stevia rebaudiana y su relación con la síntesis de glicósidos.
| Gen | Función | N° de reacción catalizada en la ruta metabólica |
|---|---|---|
| 1 al 9 | Síntesis de UDP-glicosiltransferasas | No definido |
| 10 | Síntesis de 4-(Citidina5’difosfo)-2-metil-D-eritritol 2-fosfato a partir de 4-(Citidina5’difosfo)-2-metil-D-eritritol | 4 |
| 11 | Isomerización de dimetilallil difosfato a isopentenildifosfato | 8 |
| 12 | Síntesis de geranilgeranil difosfato a partir de dimetilallil difosfato | 9 |
| 13 | Ciclación de geranil geranil pirofosfato (ggpp)en copalil pirofosfato (cpp) | 10 |
| 14 y 15 | Síntesis de (-)-kaureno por ionización dependiente de la ciclación del copalil difosfato | 11 |
| 16 | Síntesis de esteviol (ent-kaur-16-en-13-ol-19-oic ácido) por hidroxilación del ácido ent-kaurenoico (ent-kaur-16-en-19-oic ácido) con la participación de NADPH y oxígeno molecular | 13 |
| 17 | Síntesis de esteviolmonósido a partir de esteviol | 14 |
| 18 | Síntesis de esteviósido a partir de esteviobiósido | 16 |
| 19 | Síntesis de rebaudiósido A partir de esteviósido | 17 |
| 20 | No definida | No definido |
Fuente: NCBI (2014), Kumar et al. (2012).
Cuadro 3 Función de 11 sitios de regulación de la transcripción descritos en Arabidosis thaliana y sus proteínas de anclaje.
| Nombre de los sitios en cis seleccionados | Función de los sitios en cis | Factor de regulación relacionado | Accesión factor de transcripción |
|---|---|---|---|
| ABRELATERD1 (S000414) | Respuesta a la deshidratación por activación de una proteína quinasa dependiente de ácido fosfatídico | Arabidopsis thaliana 3’-phosphoinositidedependent protein kinase 1 | NM_203001.3 |
| ACGTATERD1 (S000415) | Respuesta temprana a la deshidratación por la Clp proteasa como subunidad reguladora dependiente de ATP | Arabidopsis thaliana chaperone protein ClpD | NM_124486.2 (3202 pb) |
| ARR1AT (S000454) | Regulador de respuesta por activación transcripcional mediante proteína reguladora dependiente de citoquinina | Arabidopsis thaliana AT3G16857 | AK316883.1 (2346 pb) |
| DRECRTCOREAT (S000418) | Respuesta temprana a la deshidratación por acción de una histidina quinasa y un dominio de respuesta regulatoria | Arabidopsis thaliana ethylene receptor | NM_105305.4 |
| Nombre de los sitios en cis seleccionados | Función de los sitios en cis | Factor de regulación relacionado | Accesión factor de transcripción |
| EVENINGAT (S000385) | Control circadiano de la expresión | -- | -- |
| LTRECOREATCOR15 (S000153) | Respuesta a bajas temperaturas | -- | -- |
| MYB2CONSENSUSAT (S000409) | Sitios de reconocimiento del factor de transcripción MYB que posee un dominio de unión de ADN MYB R2R3 y es conocido por regular la expresión de genes de respuesta a deshidratación y salinidad | Arabidopsis thaliana R2R3 MYB DNA binding domain transcription factor | NM_130287.2 |
| MYBCORE (S000176) | Respuesta a estrés hídrico y estímulo a auxina, etileno, ácido jasmónico, ácido salicílico y sal | Arabidopsis thaliana myb domain protein 14 | NM_128674.3 |
| MYCATERD1 (S000413) | Respuesta temprana a la deshidratación | - | - |
| MYCATRD22 (S000174) | Respuesta a la deshidratación, estrés oxidativo, ABA relacionado con síntesis de triptófano | Arabidopsis thaliana transcription factor MYC2 | NM_102998.3 |
| MYCCONSENSUSAT (S000407) | Sitios de reconocimiento del factor de transcripción bHLH de desarrollo morfológico de hoja | Arabidopsis thaliana transcription factor EGL1 mRNA | NM_001198373.1 |
Fuente: Interactome (2014), NCBI (2014), PLACE (2014).
Secuenciación y análisis de transcriptoma
Las condiciones de la muestra de ARN total a partir de muestras de hoja, previamente a su secuenciación, se resumen en una concentración de 84,6 ng/ µl, OD260/280 1.9 y RIN de 9.3.
Los datos de secuenciación se dividieron en dos librerías tipo paired-end, cada una con 28 360 329 fragmentos, con una proporción de guanina-citosina de 52,7%. El porcentaje de fragmentos con calidad Sanger superior a 30% fue de 88,81%. Del total de 56 720 658 fragmentos generados con la secuenciación, 53 998 440 fragmentos pasaron el sistema implementado con la eliminación de bases de baja calidad (27 627 790 en la librería 1 y 26 236 583 en la librería 2).
La calidad de los fragmentos de la librería 1 fue superior al 30% en su totalidad, incluyendo el rango de la desviación estándar. En la librería 2 la calidad promedio también fue superior al 30% y los valores de la desviación estándar superiores al 25% (figura 1).
Alineamiento de genes contra el transcriptoma:
El alineamiento entre los fragmentos de secuenciación correspondientes a los ocho genes de ruta metabólica de síntesis de glicósidos en S. rebaudiana se presenta en el cuadro 4.
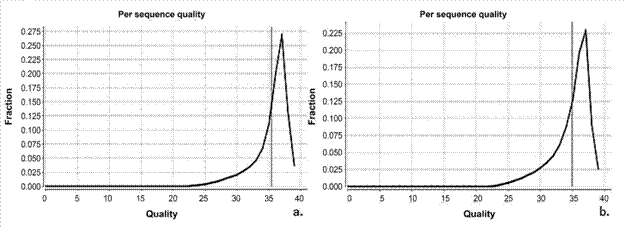
Figura 1: Distribución de la calidad a lo largo de los fragmentos amplificados de ADN copia de Stevia rebaudiana con secuencias depuradas: a. Librería 1, b. Librería 2. Fuente: RobiNA (2014).
Cuadro 4 Alineamiento de ocho genes descritos para Stevia rebaudiana contra el transcriptoma de esta misma especie.
| N° de gen | N° de nucleótidos del gen (pb) | N° de fragmentos alineados | N° mínimo de nucleótidos alineados | % de alineamiento |
|---|---|---|---|---|
| 13 | 2590 | 6 547 | 458 290 | 0.02 |
| 14 | 2792 | 170 | 11 900 | 0.00 |
| 15 | 3117 | 230 | 16 100 | 0.00 |
| 16 | 1678 | 384 | 26 880 | 0.00 |
| 17 | 1586 | 4479 | 313 530 | 0.02 |
| 18 | 1555 | 521 | 36 470 | 0.00 |
| 19 | 1616 | 1407 | 98 490 | 0.01 |
| 21 | 1396 | 1347 | 94 290 | 0.01 |
Fuente: RobiNA (2014).
Discusión
En total se identificaron 20 genes de S. rebaudiana, los cuales pudieron ser relacionados con la ruta metabólica de síntesis de glicósidos, de acuerdo con la función descrita en la base de datos del NCBI así como aquella descrita en la investigación que Kumar et al. (2012) han desarrollo, documentando comportamientos fisiológicos y describiendo genes para esta especie.
De los 20 genes de estevia descritos en las bases de datos, únicamente de cinco de ellos se conocía la región promotora, que fue analizada para determinar los posibles sitios de anclaje a ADN por parte de proteínas de regulación. En 2013 se realizó un estudio similar (Kumar & Guleria, 2012), para determinar los sitios en cis correspondientes a las cajas TATA y GATA, con la finalidad de evaluar la producción de glicósidos ante estímulos lumínicos en estevia.
En relación con las regiones promotoras descritas en S. rebaudiana, la herramienta de análisis seleccionada generó predicciones acerca de la posible ubicación de las cajas TATA, GATA y CAAT; además, también se pudo señalar la ubicación de posibles sitios de anclaje a otros factores de regulación como los factores de transcripción.
La selección de los sitios de interés se efectuó mediante una discriminación de acuerdo con su descripción para la especie modelo A. thaliana, específicamente para tejidos de hoja. Esto se debe justamente a que la complejidad de las interacciones que ocurren entre el ADN y las proteínas reguladoras de la transcripción depende de los diferentes tejidos del organismo, sus tipos celulares, estadíos de desarrollo, estados fisiológicos y condiciones ambientales y/o hormonales (Carlberg & Molár, 2014; Ceunen & Geuns, 2013; Latchman, 2003; Zhang, 2003).
Por la razón anterior es que también se escogieron sitios promotores que se relacionaran con respuestas típicas en hojas, como ante condiciones de estrés hídrico y deshidratación, principalmente porque las hojas se encargan del proceso de fotosíntesis y respiración, durante los cuales puede darse la pérdida de agua a través de los estomas (Audersik et al., 2008). De igual manera, se escogieron sitios que se vincularan con respuesta a niveles de salinidad y ataque de insectos, porque la literatura reporta investigaciones al respecto en estevia, cuyo objetivo ha sido comprender y relacionar la síntesis de compuestos bioactivos como los glicósidos, según diversos estímulos ambientales.
En este estudio, una vez establecida la función concreta de los factores de regulación seleccionados de A. thaliana, se determinó que solo tres de ellos correspondieron a factores de transcripción propiamente dichos, estos fueron proteínas con dominios MYC y MYB. Las proteínas MYB se relacionan con la respuesta de estrés de las plantas, como exposición a luz ultravioleta, heridas, estrés anaeróbico y entrada de patógenos (Singh et al., 2002). Las proteínas MYC se relacionan con la diferenciación de las células epidérmicas y patrones de tricomas, actuando como cofactores con otras proteínas (Schwechheimer et al., 1998).
Tras el final de la etapa de búsqueda de genes, se continuó con el proceso de secuenciación del transcriptoma. Si bien la estrategia para medir la expresión génica es la comparación del transcriptoma contra un genoma de referencia, muchas veces, dependiendo del organismo en estudio, esta aproximación se dificulta cuando no se cuenta con un genoma de referencia conocido o este está parcialmente descrito (Grabherr et al., 2011). Una de las estrategias propuestas para solventar este problema es la comparación de los fragmentos o los transcriptomas ensamblados de novo, contra organismos modelo filogenéticamente cercanos (Hornett & Wheat, 2012). Los niveles de expresión génica para cada unidad de ARN pueden aproximarse de acuerdo con el número de fragmentos secuenciados que se alinean con un genoma de referencia, relacionándolos directamente con niveles de abundancia (Trapnell et al., 2010).
Con el análisis de calidad de la secuenciación de estevia se eliminaron 2 722 218 fragmentos, lo que representa casi un 5% del total secuenciado. Un 5% representa un porcentaje bajo, por lo que se reafirma la idea de que la secuenciación realizada fue de alta calidad. Además se verificó que todos los fragmentos restantes presentaban una calidad promedio superior al 30% y una longitud de entre 70 y 80 bases.
En el análisis de alineación entre los ocho genes de estevia y el transcriptoma, todos los genes de ruta metabólica de síntesis de glicósidos estaban presentes en la muestra de ARN secuenciada, ya que, aunque se considera que un mapeo es válido si se genera más de un 80% de alineamiento contra un transcriptoma y que con menos de un 50% cuestiona si los fragmentos representan un transcriptoma incompleto o son contaminación de la muestra (Lin, 2013)., esto únicamente es válido cuando el mapeo se realiza contra la totalidad de un genoma, pero en este caso no, porque se trabajó con genes de entre 2000 y 3000 pb; por tanto, los bajos porcentajes de alineación son esperables y justificados.
Es decir, el resultado obtenido en esta sección fue positivo para la comprobación molecular de síntesis de compuestos bioactivos (glicósidos) en la muestra de hoja trabajada. Ahora, con base en el principio de que la expresión de un gen puede relacionarse con estos datos de alineación obtenidos, es importante notar que algunos de los genes seleccionados se encontraron menos representados en el transcriptoma, lo cual puede ser un indicador de lo que algunos autores describen como la producción controlada de glicósidos dada por ciertos genes.
Con respecto a lo expuesto con anterioridad, los genes 14, 15, 16, al alinear con un menor número de fragmentos, podría considerarse que su expresión en la muestra era menor en comparación con los demás genes. Esto concuerda con los estudios correlativos entre las enzimas de ruta, que han puesto de manifiesto la importancia de los genes denominados como SrCPPS (13), SrKS (14 y 15), SrKO en la regulación del contenido de glicósidos (Kumar et al., 2012). En esta investigación, el paso de copalil difosfato a kaureno, de kaureno a ácido kaureonico y de este último a esteviol fueron los pasos más regulados.

