











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Plasmodium vivax is a virulent ma laria parasite that widely spreads in Southeast Asia, the Americas, the Eastern Medi terranean, and the Western Pacific (Howes et al., 2016; World Health Organization, 2020). In 2019, this parasite became the dominant species that caused malaria dis ease in Southeast Asian regions, accounting for more than 50% of cases, which totaled 6.3 million (World Health Organization 2020). Furthermore, 80% of the 1.6 billion population in Southeast Asia has a risk of vivax malaria transmission (Battle & Baird, 2021). Unlike Plasmodium falciparum, this species has a unique dormant stage by forming hypnozoites. A hypnozoite is a dormant form of parasite that is activated after further infection (Dini et al., 2020; Markus, 2011 This uniqueness causes vivax malaria to be called a relapsing malaria disease.
In malaria, blood infection becomes a crucial stage to expand parasite invasion and increase its population. Inhibition at this stage probably prevents important life cycle continuation, such as gametocyte, trophozoite, and schizogony stage development. The success of Plasmodium vivax invading red blood cells depends on the ex istence of a Duffy-binding antigen protein (Howes et al., 2016). Hence, host-pathogen molecular interaction through the Duffy antigen protein during the blood stage is re quired to support the parasite invasion pro cess. One of the parasite’s proteins included in the mechanism is the Plasmodium vivax Duffy Binding Protein (PvDBP). This protein is a member of the erythrocyte binding- like (EBL) protein superfamily, consisting of 330 amino acids and twelve conserved cysteine residues (Batchelor et al., 2011; Hans et al., 2005). The PvDBP is localized in the micronemes organelle and released during the invasion process (Han et al., 2016). The PvDBP binds to Duffy antigen receptor chemokines (DARC) driven via the dimerization process (Batchelor et al., 2011). This interaction mediates merozoite internalization into red blood cells. Because of its vital role in parasite development, inhibition of PvDBP-DARC interaction may greatly contribute to parasite control.
Malaria treatment using chloroquine is still the mainstay, especially for vivax malaria in several regions (Chu & White, 2021; Ould et al., 2015; Sutanto et al., 2010). Even though multiple studies revealed that chloroquine reduces its efficacy to cure vivax malaria disease due to resistance occurrence (Ferreira et al., 2021; Getachew et al., 2015; Htun et al., 2017), other studies reported that Plasmodium vivax also resists several antimalaria drugs, including pyrimethamine and sulfadoxine (Korsinczky et al., 2004; Ratcliff et al., 2007). Therefore, the emergence of Plasmodium vivax resistance becomes a challenge in identifying novel therapeutic agents, especially from natural resources.
The study of plant metabolites to find out their potential as drug candidates or many diseases has been encouraged. Several reports demonstrated that secondary metabolites from plants show positive results in decreasing malaria parasite infection (Abdou et al., 2022; Ahmed et al., 2022; Okokon et al., 2022). The Zingiberaceae plant family is one of the popular plants widely used in traditional medicine to cure some diseases. Almost all parts of this plant potentially contain beneficial bioactive compounds. Therefore, the evaluation of its potential as a drug candidate, especially for vivax malaria, must be expanded.
In this study, we use a bioinformatics approach to predict interactions between bioactive compounds from the Zingiberaceae plant family targeting the PvDBP. We generally use two types of analysis, including molecular docking and dynamic simulation. We analyze compounds-protein interaction types and strength by using molecular docking, whereas molecular dynamic simulation is used to analyze the protein behavior of PvDBP-ligand complexes during simulation time.
Material and Methodology
Protein Target Retrieval A PvDBP 3D structure file (PDB ID: 6R2S) in PDB format was obtained from RSCB PDB database (https://www.rscb.org/). Before entering the docking process, we removed all native ligands that bind into protein using PyMOL v.2.5.2 software (https://www.pymol.org).
Ligand Selection and Preparation Approximately 200 bioactive compound data were collected from 15 Zingiberaceae plant species, as in our previous study (Heikal et al., 2023). The data were referred from several previous reports. Moreover, bioactive compound SDF files were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov), in accordance with our previous studies (Hidayatullah et al., 2022; Hidayatullah et al., 2021; Putra, 2018; Putra et al., 2017). To examine compounds’ physical and chemical properties, we performed a drug-like test using the Lipinski rule of five web server (http://www.scfbio-iitd. res.in/software/drugdesign/lipinski. jsp). There are about five indicators used as key factors in determining the drug-likeness of ligands, including hydrogen bond donor, hydrogen bond acceptor, molecular weight, lipophilicity, and molar reactivity (Lipinski, 2004).
Molecular Docking Screening Molecular docking procedures were followed in our previous studies (Widiastuti et al., 2023; Hidayatullah et al., 2021; Putra & Rifa’I, 2020; Putra et al., 2020). The PyRx integrated with autodock vina (https://pyrx.sourceforge. io/) was used for the docking screening process. After 137 compounds passed drug-like testing, they were minimized and converted into PDBQT format. Chloroquine as vivax malaria was used as control. The grid size used was X: 66.2301, Y:-104.93, Z:-9.2953, and the coordinate dimension (Angstrom) was X: 84.4322, Y: 58.3046, Z: 57.4419.
Data Visualization We performed two types of visualization processes (Putra et al., 2023; Hidayatullah et al., 2021; Putra et al., 2019). The top two compounds + control - PvDBP complexes were visualized to determine the type of interaction and binding region. We used LigPlot+ v.2.2 (https://www.ebi.ac.uk/thornton-srv/software/LigPlus) for 2D visualization and PyMOL v.2.5.2 (https://www.pymol.org) for 3D visualization.
Molecular Dynamics Simulation We performed a molecular dynamic simulation of three PvDBP-ligand complexes in 1000 picoseconds simulation time similar to our previous simulation setting (Maslikah & Putra, 2024; Hidayatullah et al., 2023; Sari et al., 2023; Ashar et al., 2023). The complexes were prepared ackcal/mol) and pinostrobin (CID: 6950539) is the second (-9.2 kcal/mol). Around 80% of both binding residues are DARC binding residues. In contrast, chloroquine as control has a higher binding energy value (-5.5 kcal/mol) with 50% residues are DARC binding residues. We predict that the attachment of two potential compounds and control can disturb PvDBPDARC molecular interaction during merozoite invasion (Figure 1).
Pharmacokinetics Prediction To predict the adsorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the selected compounds, we performed pharmacokinetics prediction using the pkCSM webserver (https://biosig.lab.uq.edu.au/ pkcsm/). The prediction of the pharmacokinetics parameters is important in drug discovery due to its contribution to initial results that could be referred for further drug development.
Analysis and Results
About 170 amino acids were identified as DARC binding site residues of PvDBP. All three compounds partially bind to the crucial binding residue (Table 1). 5.7-Dihyroxyflavanone (CID: 238782) has the lowest binding energy value (-9.3 kcal/mol) and pinostrobin (CID: 6950539)
is the second (-9.2 kcal/mol). Around 80% of both binding residues are DARC binding residues. In contrast, chloroquine as control has a higher binding energy value (-5.5 kcal/mol) with 50% residues are DARC binding residues. We predict that the attachment of two potential compounds and control can disturb PvDBPDARC molecular interaction during merozoite invasion (Figure 1).
Table 1 Binding residue details. Bold indicates Duffy Antigen Membran Chemokines (DARC) binding residue.
| Compounds | Binding Residue |
| 5,7-Dehydroxyflavanone | Thr259, Phe267, Asn256, Asp258, Tyr271, Arg268, Ile335, Asn331, Ser334, Leu253 |
| Pinostrombin | Ile335, Leu253, Tyr271, Phe267, Thr259, Asn256, Ser334, Asn331, Arg268, Asp258 |
| Chloroquine | Asp483, Phe485, Glu493, Phe490, Val408, Leu404, Pro405, Val401 |
Two-dimensional visualization pre- and post-MD simulation shows there are alterations of all PvDBP-ligand bonding before and after simulation (Figure 2). Chloroquine lost around 75% of its binding residue towards PvDBP and just retained binding with one DARC residue (Leu404). On the other hand, 5,7-Dyhydroxyflavanone lost its interaction with Phe267 and Arg268 and made new bonds with Asn260. Furthermore, Pinostrobin lost its interaction with two residues (Asn256, Thr259) without making new bonds with other residues.
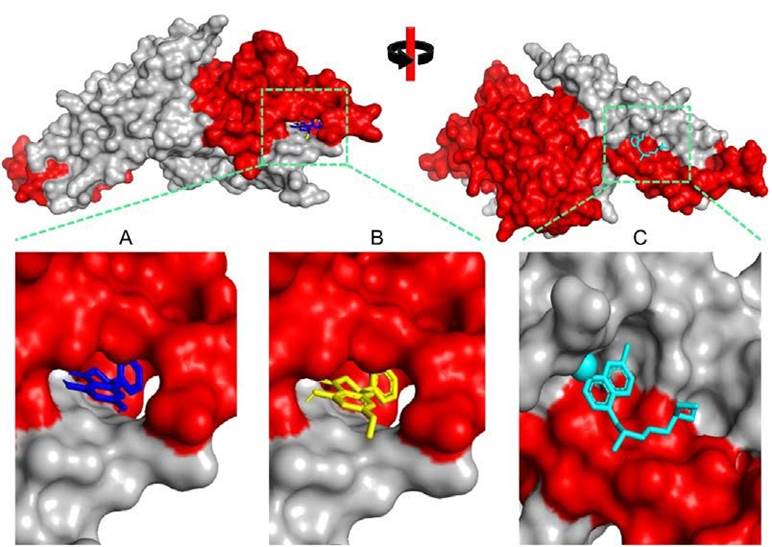
Figure 1 Three-dimensional visualization of Plasmodium vivax Duffy Binding Protein (PvDBP)-ligand complexes. A) 5,7-Dihydroxyflavanonne, B) Pinostrombin, and C) Chloroquine. The red color indicates the DARC binding region.
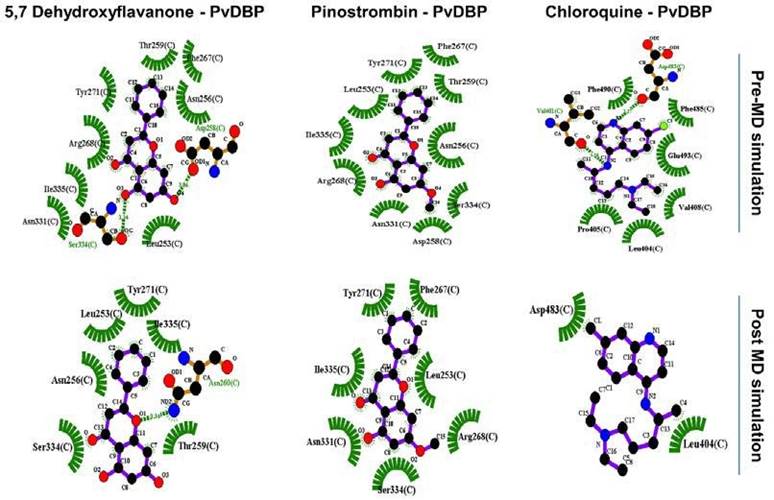
Figure 2 PvDBP-ligand two dimensional visualization before and after 1000 ps molecular dynamic (MD) simulation.
Intramolecular hydrogen bonds do not change significantly over 1000 ps simulation time. PvDBP-5,7-dihydroxyflavanone and PvDBP-chloroquine reach 280 H-bonds in 200 and within 750-800 picoseconds, respec tively. On the other hand, the protein-solvent H-bond plot shows that PvDBP-5,7-Dehydroxyflavanone lost its 40 H-bond within 450-500 ps. However, overall, the trends of all three complexes look stable.
The RMSD graph (Figure 3) shows that protein backbones are relatively steady within the 1000 ps simulation after reaching the equilibrium point. The PvDBP - 5,7-dihydroxyflavanone complex has an RMSD value of 1.586 Å, ranging from 0.493 Å to 2.002 Å. Then, the PvDBP-Pi nostrobin shows an RMSD value of 1.561 Å, with a maximum score of 1.98 Å and a minimum score of 0.48 Å. Meanwhile, the PvDBP-Chloroquine complex has the highest RMSD score over the 1000 ps simula tion 2.302 Å.
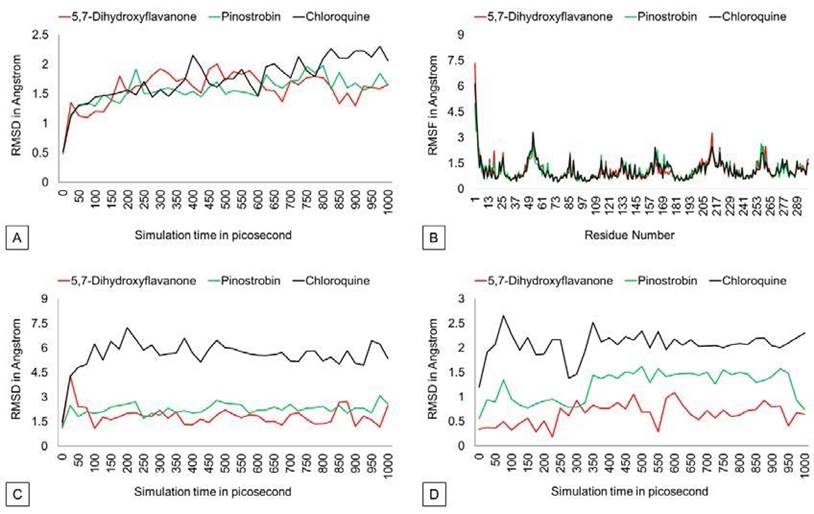
The RMSF graph (Figure 3) shows that all three complexes relatively have similar stability over simulation. Highest peaks of 3 complexes were reached by PvDBP-Chloroquine in between 49-61 residues and PvDBP-5,7-dihydroxyflavanone in between 205-217 residues. RMSD Ligand conformation score shows PvDBP-5,7-Dihidroxyflavanone has the lowest value of 0.651Å, whereas PvDBP-chloroquine is the highest. In addition, PvDBP-Chloroquine has the largest value of ligand movement score of 16280.572 Å, which reaches its highest peak of 16712.116Å.
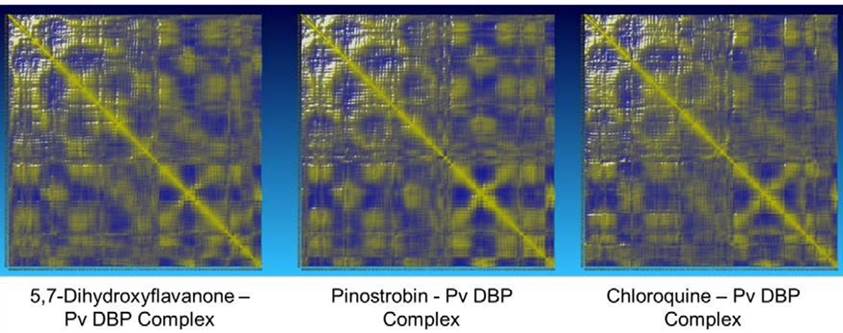
The potential energy value of all protein-ligand complexes is relatively flat over simulation (Figure 4). It indicates that PvDBP-ligand complexes maintain their stability to store potential energy. On the other hand, Protein-ligand binding also does not affect protein expansion. These three complexes reach nearly conformable trends after reaching 400 ps. PvDBP-Chloroquine reached maximum SASA score 16712.116 Å2 at 250 ps. The dynamic cross-correlation maps of three protein-ligand complexes (Figure 5) reveal no significant distinction about the at-om’s physical motions. The blue color stands for anti-correlated motions, while yellow represents correlated motions. Thick color stands for high flexibility of protein residues.
PvDBP localized in micronemes or-ganelle and presented in Plasmodium vivax surface when merozoite starts the invasion process (Adams et al., 1990; Fraser et al., 1997). PvDBP interacts with DARC through binding with central 170 amino acids in PvDBP region II (RII-PvDBP) (Hans et al., 2005; Singh et al., 2003). PvDBP-DARC
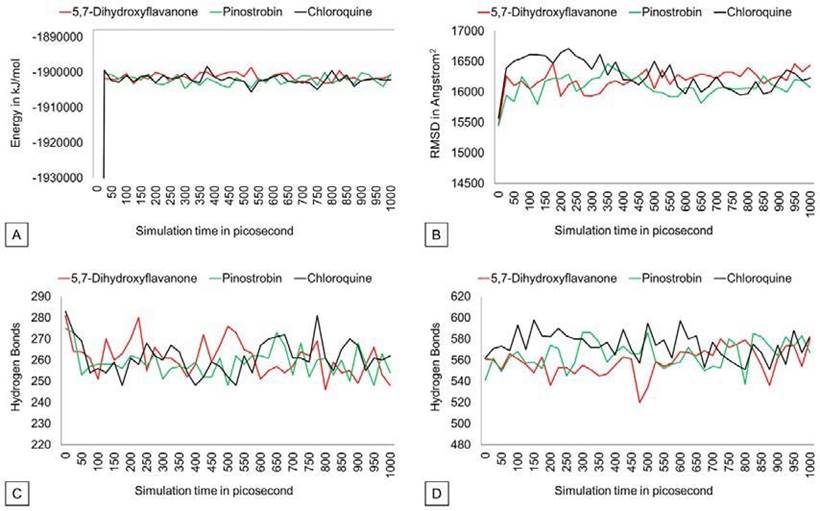
Figure 4 Potential energy, SASA, hydrogen bond (intramolecular), Hydrogen bond solvent (solute) graph.
molecular interaction is driven by the dimerization process of PvDBP homodimer (Batchelor et al., 2011). Interaction between two molecules of PvDBP resulting in DARC binding groove. In this investigation, all three compounds predicted partially bind into crucial binding residues of DARC.
5,7-dihydroxyflavanone (Pinocembrin) is a flavonoid compound found in the rhizome of Kaempferia rotunda (Atun et al., 2013). The lowest binding energy value towards PvDBP indicates this compound is predicted to have a stronger tendency to incorporate with PvDBP than control. Almost all binding of this compound is located in subdomain 2 of RII-PvDBP. Subdomain 2 of RII-PvDBP is bounded by all DARC-interacted residues (Batchelor et al., 2014). We predicted that the binding stability of 5,7-dihydroxyflavanone was relatively strong with retained bonding with 60% residues and made novel interaction with one DARC binding residue during simulation time. In silico and in vivo studies reveal that pinocembrin and its analog show great antiplasmodial activity (Melaku et al., 2022).
The second compound that may have potential as a drug for malaria is pinostrobin. Pinostrobin is a flavonoid compound that is found in Bosenbergia rotunda (Kanchanapiboon et al., 2020; Morikawa et al., 2008; Tewtrakul, 2005). The binding strength of this compound is not much different from 5,7-dihydroxyflavanone. This compound is predicted to bind to subdomain-1 and subdomain-2 of RII-PvDBP. This compound also binds to Tyr271, whereas this residue plays a role in the PvDBP-DARC dimer interface (Batchelor et al., 2014). Moreover, this pinostrobin-PvDBP interaction is also predicted to disturb the PvDBP-DARC second interface process through bonding with Phe267. Phe267 of RII-PvDBP is required for the DARC binding process and recognized by neutralizing antibodies (Batchelor et al., 2014; Chootong et al., 2010).
Merozoite invasion of red blood cells is required for parasite further development. The parasites digest hemoglobin contained in red blood cells to support their growth (Moore et al., 2006; Pishchany & Skaar, 2012). The PvDBP-compounds interaction in the crucial residues revealed that the DARC-PvDBP association can be disturbed. It affects the internalization process of merozoites in the early blood stage cycle. Inhibition of merozoite internalization possibly controls parasite development extension, throphozoite, schizont, and gametocyte development.
In molecular dynamic analysis, we predicted that 5,7-dihydroxyflavanone-PvDBP and pinostrobin-PvDBP complexes have greater stability than the control. The RMSD value < 2 Å indicates that all three complexes are remarkably stable during simulation time (Castro-Alvarez et al., 2017). Furthermore, protein-ligand binding can lead to protein conformational changes like subdomain and protein backbone movement (Frimurer et al., 2003). Study results reveal that interaction between the two bioactive compounds did not result in a significant conformational alteration of PvDBP. In the RMSF analysis, all three compounds have quite similar effects on binding residue distance. RMSF analysis was used to analyze protein residue mobility during protein-ligand interaction (Zhu et al., 2017). The result indicates that protein-ligand interactions have almost similar binding residue distance fluctuation within the simulation.
All three complexes show stable trends in the SASA value plot. It revealed that ligand-protein binding does not affect the protein’s surface expansion due to the unfolding process (Candotti et al., 2013) Intramolecular hydrogen bonds are required to retain protein structure by maintaining protein secondary structure (Gao et al., 2015; Hubbard & Kamran Haider, 2010).
An increase in intramolecular H-bonds indicates that the protein committed a dena turation process by interacting with solvent hydrogen atoms. The number of intramolecular H-bonds of three complexes is relatively stable. This means that the protein-ligand complex did not make significant H-bond number changes. DCCM analysis was used to observe correlative motions of the protein atom during MD simulation (Arnold & Ornstein, 1997; Kasahara et al., 2014). All three complexes convey almost similar flexibility among protein residues.
Pharmacokinetics properties predic-tion is important to measure the capabilities of the selected compound to be processed, delivered, and excreted inside the human body. Five criteria were used to determine the pharmacokinetics properties of the selected compound by docking studies (Figure 6). Both top two compounds exhibit good adsorption properties with relatively the same water solubility score, ranging from -3.538 to -4.249. The top two compounds possess higher intestinal absorption compared to chloroquine as a control. CaCo2 permeability indicates that the compounds are easily adsorbed to human colorectal adenocarcinoma cells with the value > 0.90 Sun et al., 2017). The top two compounds have a low VDss value, while the chloroquine is high (1.332). The findings suggest that the compound is widely distributed in plasma rather than tissue. Chloroquine and 5,7-dihydroxyflavanone were predicted to be readily distributed to the brain due to its logBB score > 0.3. However, among the three compounds, they have a low ability to pen-etrate the central nervous system. In terms of metabolism properties, the capability of
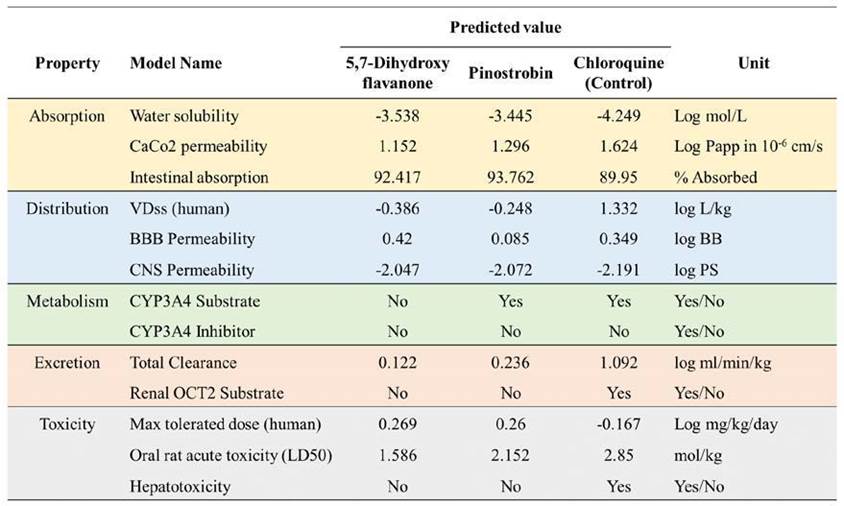
the protein to affect Cytochrome P450 function is considered. This protein acts in the activation and deactivation process of the drugs. There are no compounds that possess the ability as a CYP3A inhibitor. Moreover, only 5,7-dihydroxyflavanone was predicted to influence CYP3A function as a substrate. Chloroquine is predicted to have a higher total clearance score. This parameter is utilized to determine the dosing rate of the compounds to make steady-state concentrations. In toxicity prediction, we found that the two bioactive compounds have less toxicity than chloroquine. These compounds have a higher maximum tolerated dose (human) and lower LD50 scores. Furthermore, 5,7-dihydroxyflavanone and pinostrobin were predicted to not have hepatotoxicity properties. Toxicity prediction assists in reducing experimental costs and time while also addressing ethical issues by considerably lowering animal and clinical testing requirements (Sharma et al., 2023; Cavasotto & Scardino, 2022).
Conclusions
The present study aims to screen numerous Zingiberaceae bioactive compounds as potential inhibitors targetting PvDBP. We found that two compounds (5,7-dihydroxyflavanone and pinostrobin) may prevent PvDBP and DARC interaction due to their strong binding towards DARC binding residues. This research is the first investigation of ligand screening that targets PvDBP as a drug target. Therefore, the findings are important as baseline information for further natural chemistry-based drug development against vivax malaria. Nonetheless, this prediction still requires validation through in vitro and in vivo experiments.
Funding
This study was funded by PNBP Universitas Negeri Malang with contract numbers 5.3.619/UN32.14.1/LT/2021 and 5.3.517/UN32.14.1/LT/2021.
Acknowledgments
The authors thank the Laboratory of Biocomputation, Department of Biology, Brawijaya University, for supporting this study.
Author contribution statement
All the authors declare that the final version of this paper was read and approved.
The total contribution percentage for the conceptualization, preparation, and correction of this paper was as follows: M.F.H. & W.E.P. 70%; S., A.H., D.W. & M.L 30%.

