









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
1. Introducción
La población infantil con discapacidad intelectual (DI) debe ser sometida a diversos estudios tanto genéticos como no genéticos desde sus primeros años de vida para determinar el posible origen. Entre los más relevantes, se encuentra el análisis citogenético o cariotipo, pues las aberraciones cromosómicas son una causa importante de dicha condición. Por ejemplo, la trisomía 21 se puede detectar fácilmente en un cariotipo, sin embargo, diversos estudios han evidenciado la necesidad de evaluar el genoma, de forma que, también se puedan hallar aberraciones submicroscópicas, especialmente en las regiones subteloméricas, por ser propensas a sufrir rearreglos cromosómicos asociados con la DI.
De ahí la trascendencia de implementar en el país estrategias diagnósticas al servicio de este propósito y disponibles para pacientes costarricenses afectados y para el personal médico que les atiende. En ese sentido, este trabajo contribuye a ampliar el repertorio de pruebas diagnósticas alrededor de esta temática.
2. Referente teórico
La DI se caracteriza por presentar limitaciones intelectuales y adaptativas y aparece antes de los 18 años, (AAIDD, s.f.; Vissers et al., 2016). Su prevalencia en la población mundial es del 2% al 3 %; no obstante, a pesar de los avances en cuanto a técnicas diagnósticas, alrededor de la mitad de los casos (50 % - 60 %) siguen sin explicación a su causa (Medina et al., 2014; Vissers et al., 2016).
Dentro de las condiciones genéticas de DI, se incluyen aquellas donde prevalece una mutación en un único gen (monogénicas) (Hamdam et al., 2014), alteraciones en los mecanismos epigenéticos (Ramsden et al., 2010) y aberraciones cromosómicas (Tomac et al., 2017). En años recientes, las variantes en el número de copias se catalogaron como un nuevo factor genético, lo cual podría justificar una proporción importante de los casos clasificados como idiopáticos (de origen desconocido). Estas alteraciones se conocen como aberraciones cromosómicas submicroscópicas, porque comprenden deleciones o duplicaciones muy pequeñas (<5Mb) que no pueden ser detectadas mediante técnicas de citogenética convencional. Se ha estimado que entre el 5 % y el 7 % de los casos de DI idiopático conllevan alteraciones subteloméricas submicroscópicas (Mohan et al., 2016; Meyyazhagan et al., 2021).
Los subtelómeros son regiones ricas en genes que pueden diferir en cuanto a tamaño y contenido de información y esto aumenta la probabilidad de apareamientos erróneos durante la meiosis (Mohan et al., 2016). Debido a lo anterior, se han utilizado diferentes técnicas moleculares para el estudio de las microdeleciones y las microduplicaciones, entre ellas, la hibridación in situ fluorescente (FISH, del inglés fluorescence in situ hybridization) (Hyun-Kyung et al., 2008; Karnebeek et al., 2002; Mohan et al., 2016; Meyyazhagan et al., 2021), la hibridación genómica comparativa (CGH, del inglés comparative genomichybridization) (Kirchhoff et al., 2005), la reacción en cadena de la polimerasa cuantitativa (qPCR, del inglés quantitative polymerase chain reaction) (Auber et al., 2009) y la amplificación de sondas dependientes de ligación múltiple (MLPA, del inglés multiplex ligation-dependent probe amplification) (Ahn et al., 2007; Boggula et al., 2014; Medina et al., 2014; Rooms et al., 2006). De todas estas, la MLPA brinda la mejor relación costo-beneficio, además, ya ha sido reconocida como una técnica confiable y sensible para detectar reacomodos subteloméricos, incluyendo deleciones y duplicaciones (Ortiz et al., 2021; Santa María et al., 2016).
En Costa Rica, desde hace varias décadas, se diagnostican síndromes genéticos habitualmente responsables de DI, como los causados por aberraciones cromosómicas (aneuploidías o aberraciones de tipo estructural) (Castro et al., 2011) y el del cromosoma X frágil (SXF) (Vindas-Smith et al., 2011). En el caso de los desbalances subteloméricos submicroscópicos, hasta ahora era imposible identificarlos en nuestro país, lo que dificultaba el abordaje de los pacientes e impedía una estimación de su frecuencia. Pese a los esfuerzos por determinar las causas de DI en la población costarricense, siempre permanece un porcentaje con DI idiopática; en este último podrían ocurrir aberraciones cromosómicas submicroscópicas, de las cuales las subteloméricas tendrían una proporción significativa.
Con base en lo expuesto, este proyecto pretendió determinar la incidencia de aberraciones cromosómicas subteloméricas submicroscópicas mediante la aplicación de la técnica MLPA en una población infantil costarricense con DI idiopática.
3. Metodología
3.1 Enfoque
Este trabajo tuvo un enfoque cualitativo, observacional y transversal con alcance descriptivo, ya que se realizó la búsqueda de aberraciones cromosómicas en una población seleccionada de personas con DI idiopática.
3.2 Población del estudio
La población de estudio estuvo constituida por 77 individuos con una DI idiopática. A todos se les tomó una muestra de sangre periférica, a fin de detectar aberraciones cromosómicas subteloméricas submicroscópicas por medio de MLPA. Del total, 49 fueron remitidos al Instituto de Investigaciones en Salud de la Universidad de Costa Rica (INISA-UCR) para el diagnóstico molecular del SXF indicado por el personal médico, a raíz del resultado negativo sobre la mutación responsable de esta enfermedad. Los restantes 28 eran pacientes con DI idiopática reclutados de centros educativos de enseñanza especial (Vindas-Smith et al., 2011) y con un resultado genético negativo para el SXF.
El flujo de trabajo consistió en efectuar un cariotipo a partir del cultivo celular de sangre periférica, con el propósito de identificar aberraciones cromosómicas en aquellos casos con material biológico disponible (n = 32 del total). Si el cariotipo resultó normal o no informativo, se procedió a extraer el ADN para la prueba de MLPA. En el resto de participantes, se aplicó únicamente la MLPA (n = 45 del total). De uno de ellos, más adelante referenciado como Caso 1, se obtuvo material biológico para el cariotipo de forma posterior al análisis con MLPA, de tal modo, en total se realizaron 33 cariotipos.
En torno a la recolección de las muestras, se contó con el consentimiento informado, aprobado por el Comité Ético-Científico de la Universidad de Costa Rica y firmado por cada paciente o por su representante legal.
3.3 Cultivo Celular
Se practicaron 33 cultivos celulares, provenientes de sangre periférica, a través de la inoculación de cinco gotas de sangre en medio de cultivo completo (PB-MAX de GIBCO), seguidamente, se incubaron por 72 horas. Una vez transcurrido ese tiempo, se realizó el arresto mitótico con Colcemid (GIBCO), el choque hipotónico con KCl 0,075 M y la fijación con una solución de Carnoy (3:1 metanol:ácido acético).
3.4 Análisis cromosómico
Las células recuperadas después de la centrifugación fueron resuspendidas y se extendió el material sobre una lámina de portaobjetos. Se completó el conteo cromosómico en al menos 20 células y se analizaron mínimo tres metafases banda a banda para cada caso. Ante la sospecha de mosaicismo, se estudiaron, por lo menos, 40 células. En este análisis se utilizó bandeo G por tripsina y giemsa (bandas GTG).
3.5 Extracción de ADN
El ADN se extrajo desde linfocitos de sangre periférica, usando el método de fenol-cloroformo basado en el protocolo de Strauss (2001), con modificaciones o con ayuda del kit QIAamp DNA Mini Kit (QIAGEN).
3.6 MLPA
Se recurrió al kit SALSA MLPA P070 Human Telomere-5 probemix (MRC-Holland, The Netherlands) para trabajar las 77 muestras. Los genes diana de las sondas de este kit se pueden revisar en www.mrc-holland.com. En referencia a la MLPA, se siguieron las instrucciones del fabricante con pequeñas modificaciones: se agregaron 4 μL de la muestra de ADN + 1 μL de Tris-HCl 50 mM (pH = 8,5). En la amplificación de las sondas que fueron ligadas, se acató el procedimiento sugerido por el fabricante.
Asimismo, en cada experimento se incluyó una muestra blanco (sin ADN) y se aplicaron de tres a cinco controles consistentes en ADN de personas sin DI, extraído por el mismo método que las muestras. Los productos de PCR fueron evaluados en un secuenciador ABI 3730XL (Applied Biosystems) de la compañía Macrogen Inc., empleando la siguiente mezcla: 0,5 μL de producto de PCR, 0,2 μL del marcador molecular LIZ-500 (Applied Biosystems) y 9,5 μL de formamida. La electroforesis contempló las indicaciones de voltaje de inyección: 1,4 kV; tiempo de inyección: 15 s; voltaje de corrida: 10 kV; tiempo de corrida: 2500 s. Los electroferogramas se analizaron utilizando el programa Peak Scanner (Thermo FisherScientific).
3.7 Análisis de datos
Para la MLPA, los datos se estudiaron con el programa Coffalyser (MRC-Holland) y, en adición, fueron corroborados en una hoja de cálculo con el programa Excel (Microsoft Office), con el propósito, en ambos casos, de obtener un cociente de dosificación (CD), donde CD = ((intensidad de fluorescencia de la sonda analizada/intensidad de fluorescencia de cada una de las otras sondas)] / ((intensidad de fluorescencia de la sonda del control/intensidad de fluorescencia de cada una de las otras sondas del control)]. El CD permite comparar con los controles, de manera que, si una persona presenta dos copias de la sonda analizada, se espera una razón 1:1 (CD cercano a 1), en las microdeleciones, se espera un CD cercano a 0,5 y en las microduplicaciones, uno aproximado a 1,5.
3.8 Confirmación de casos positivos
Los resultados positivos para la MLPA se confirmaron con el kit SALSA MLPA P249 Human Telomere-8 (MRC-Holland) o el SALSA MLPA P365 Human Telomere-14 (MRC-Holland), los cuales contienen varias sondas para el subtelómero de interés en cada caso particular (ver adelante).
4. Resultados
4.1 Frecuencia de aberraciones cromosómicas
Se calculó la frecuencia de aberraciones cromosómicas encontradas en la muestra bajo estudio. Los resultados anormales para la MLPA se presentaron en dos casos, aunque el cariotipo posterior en uno de ellos indicó que este no era submicroscópico; por consiguiente, el porcentaje de aberraciones cromosómicas submicroscópicas exclusivamente fue 1/70 (1,4 %) y el de aberraciones cromosómicas detectables al microscopio en el grupo de estudio fue 1/33 (3,0 %) (ver abajo).
4.2 Análisis cromosómico
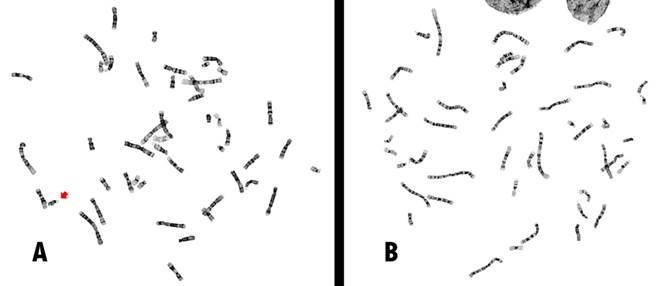
Se logró obtener el cariotipo en cada una de las 33 muestras incluidas para esta prueba. En 32 casos el cariotipo fue normal, de los cuales, 12 correspondieron a mujeres (46,XX) y 20 a hombres (46,XY). En un único caso de un varón se detectó una aberración cromosómica, cuyo cariotipo resultó ser 47,XY,+mar(32]/46,XY(11] (figura 1). Este será referido como Caso 1.
4.3 Análisis de la MLPA
De las 77 muestras de ADN de sangre periférica revisadas con la MLPA, 70 fueron concluyentes (91 %), de las cuales, 68 mostraron un resultado normal (97 %). En las dos restantes se constató una alteración en cada probando que hacía suponer la presencia de una microduplicación en cada una.
En el primer probando (Caso 1) la alteración se halló en el subtelómero 15q, en la región cercana al centrómero (llamada 15p
(6]) (figura 2). El análisis del cariotipo a posteriori descartó que se tratara de una microduplicación, sino que, el resultado anormal se debió a la presencia del cromosoma marcador. Con la prueba de MLPA fue posible corroborar que este corresponde a material genético del cromosoma 15. En el segundo probando, se observó una microduplicación en el subtelómero 17p. De ahora en adelante este será nombrado como el Caso 2 (figura 3).
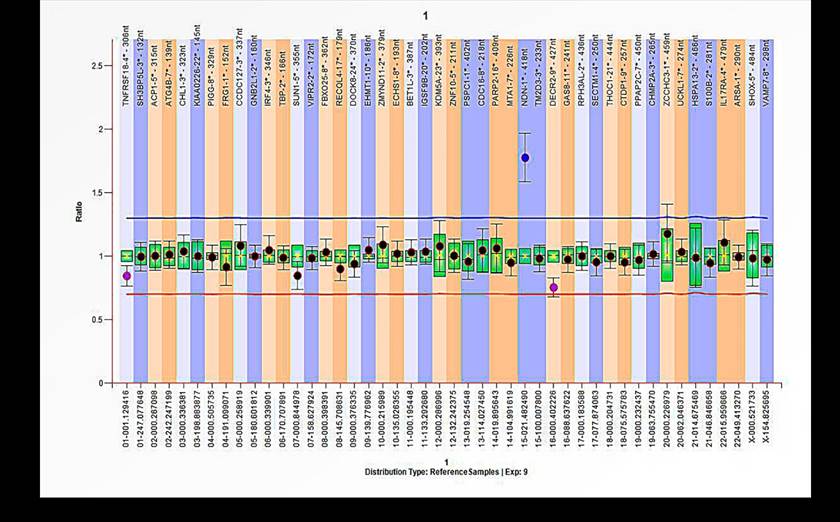
Nota. Resultado con el kit SALSA P070 donde se observa que el CD para la sonda 15p presenta un valor anormal, lo que indica la presencia de copias extra de esta región. Esta sonda corresponde al gen NDN.
Figura 2 Resultado positivo de la MLPA para el Caso 1
4.4 Análisis confirmatorios
Con respecto al Caso 1, mediante el uso del kit confirmatorio SALSA MLPA P365 Human Telomere-14, se logró determinar que los genes MKRN3, MAGEL2 y SNRPN, ubicados en la región 15q11.2, se encuentran duplicados en este probando (figura 4); así se estableció que este marcador incluye al menos dicha banda. A efectos de saber si este hallazgo correspondía a una aberración cromosómica heredada o de novo, se extrajo una muestra de sangre de los padres del probando y se aplicaron las mismas pruebas. En ambos padres, tanto el cariotipo como la MLPA resultaron normales (datos no mostrados), por tanto, la alteración genética en el probando fue de novo.
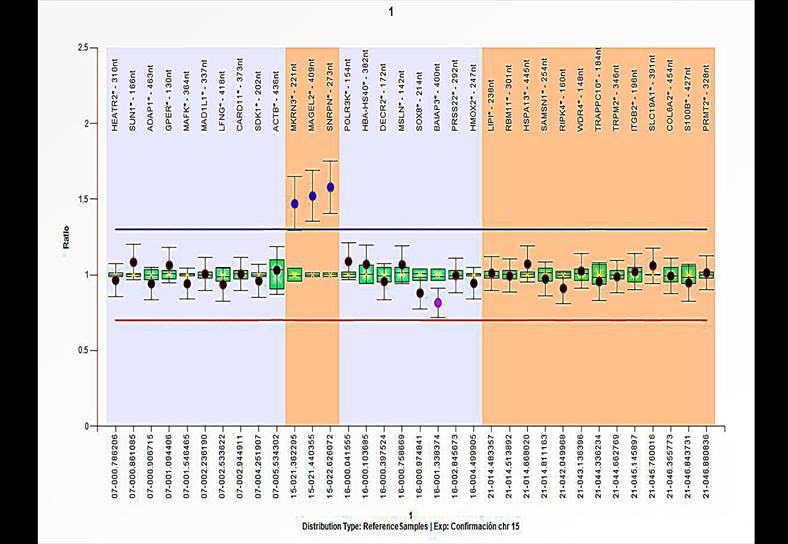
Nota. Este kit contiene tres sondas para esta región, que corresponden a los genes MKRN3, MAGEL2, SNRPN, respectivamente. Al evidenciar cocientes de dosificación elevados, se confirma el resultado.
Figura 4 Resultado positivo con el kit confirmatorio SALSA P365 para el Caso 1
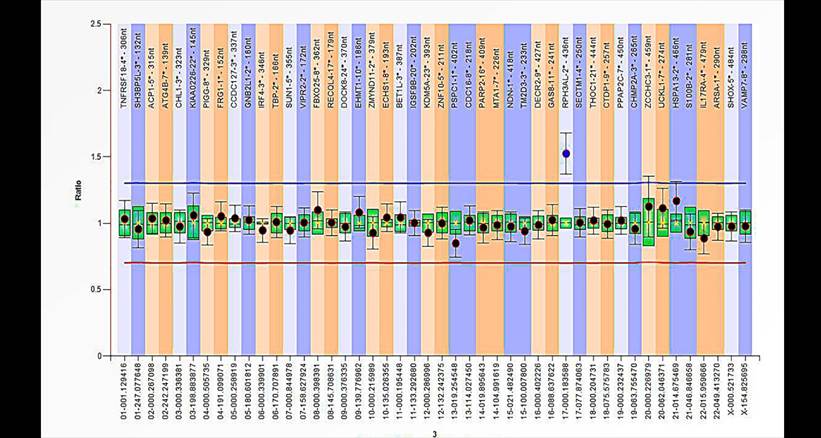
Con el propósito de comprobar el hallazgo en el Caso 2, se usó el kit SALSA MLPA P249 Human Telomere-8 para esta región y, efectivamente, el gen RPH3AL se encontraba duplicado (figura 5). En este no fue posible contactar a los padres, entonces, se desconoce si esta alteración es de novo o heredada.
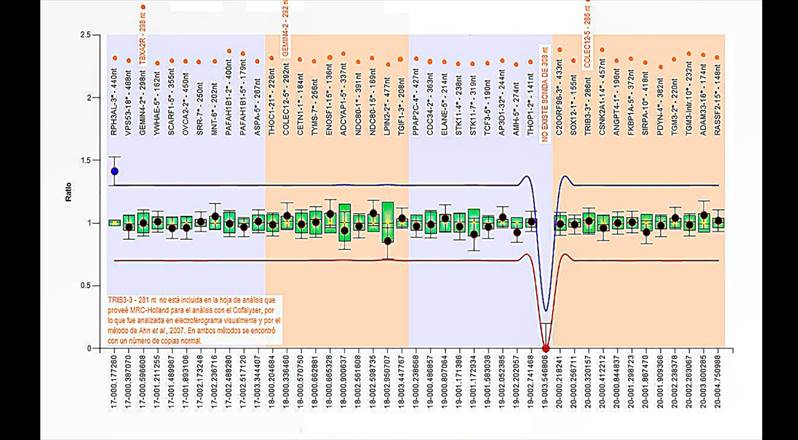
Nota. Este kit ofrece 11 sondas para la región 17p. Únicamente la sonda más distal presenta un valor alterado y responde al gen RPH3AL, lo cual confirma las copias extra de este. La hoja de análisis de Coffalyser para este kit tiene un error, por eso, los nombres correctos de los genes se visualizan en anaranjado sobre el nombre provisto por el programa (ver leyenda en letras naranja).
Figura 5 Resultado positivo con el kit confirmatorio SALSA P365 para el Caso 2
5. Discusión
5.1 Frecuencia de aberraciones cromosómicas subteloméricas submicroscópicas
En la población de estudio del presente trabajo se observó que la frecuencia de aberraciones subteloméricas submicroscópicas (1,4 %) era menor a la reportada en otros estudios (Ahn et al., 2007; Charalsawadi et al., 2016; Koolen et al., 2004); no obstante, algunos grupos han reportado prevalencias similares a las aquí descritas (Hyun-Kyung et al., 2008; Pickard et al., 2004) o inclusive menores (Karnebeek van et al., 2002). Estudios a gran escala han calculado que la aparición real de aberraciones cromosómicas submicroscópicas en los subtelómeros en personas con DI idiopática es cercana al 2,5 % (Ballif et al., 2007; Ravnanet al., 2006).
Algunos factores que dan cuenta de las diferencias surgidas de los estudios recaen en las diferentes técnicas utilizadas, si se confirma la anomalía encontrada o no, si se excluyen posibles variantes genéticas y, sobre todo, los criterios de selección de las personas (Charalsawadi et al., 2016; Hyun-Kyung et al., 2008; Karnebeek et al., 2002; Mohan et al., 2016). En este trabajo no se contempló ningún criterio de inclusión más que el diagnóstico de DI idiopática y se planteó que las altas frecuencias de aberraciones submicroscópicas en los primeros estudios se deben a una historia familiar positiva de DI, lo cual podría ser un sesgo a favor de encontrar dichas alteraciones (Karnebeeket al., 2002). Igualmente, la incidencia de las aberraciones podría variar dependiendo del grado de DI (Hyun-Kyung et al., 2008).
Según Breman y Stankiewicz (2021), la técnica de microarreglos ha desplazado al cariotipo como el primer método de elección para el diagnóstico de desórdenes del desarrollo, aunque tiene un costo muy elevado; por ende, el estudio citogenético sigue siendo importante para el análisis genético y genómico y en la actualidad puede considerarse como una técnica complementaria en muchos escenarios diagnósticos, especialmente en países donde no se cuenta con la técnica de microarreglos. Por esta razón, otros métodos de citogenética molecular sensitivas, como FISH (Mohan et al., 2016) o MLPA (Boggula et al., 2014), se recomiendan junto con el cariotipo, cuando no sean asequibles los microarreglos. En ese sentido, con este estudio se sustentó la importancia del cariotipo en el Caso 1 (posterior a la MLPA), pues se logró identificar que se trataba de un cromosoma extra estructuralmente anormal bisatelítico en mosaico y no una microduplicación.
5.2 Análisis del Caso 1
El caso con cariotipo 47,XY,+mar/46,XY corresponde a un adolescente varón de 14 años al momento del análisis. Entre las características clínicas detectadas por el personal médico se citan: trastorno de conducta, hiperactividad, escaso contacto visual y retraso del desarrollo psicomotor, además de facciones como orejas grandes y paladar ojival; con esto, se diagnosticó un posible caso de autismo o de síndrome de Asperger. El paciente fue remitido al INISA para ser evaluado mediante diagnóstico molecular para el SXF, sin embargo, el resultado fue negativo.
Conforme a lo anterior, se incluyó en este estudio, se le realizó la MLPA y, posteriormente, un análisis de cariotipo, en este último se encontró el cromosoma marcador extra. Los cromosomas marcadores poseen estructuras anormales y no pueden ser identificados solo con técnicas de citogenética convencional (Sun et al., 2020). Los cromosomas extra estructuralmente anormales (CEEA), es decir, presentes como material adicional, tienen una incidencia del 0,02 % en los recién nacidos vivos y son 4 veces más comunes en personas subfértiles y 10 veces más en personas con DI, si se comparan con la población general (DiStefano et al., 2020).
La mayoría de los CEEA se originan a partir de los cromosomas acrocéntricos y los más frecuentes son los derivados del cromosoma 15 (DiStefano et al., 2020), los cuales se relacionan con edad materna avanzada al momento de la concepción (Kurtas et al., 2019; Wu et al., 2020). Estos usualmente se derivan de una duplicación invertida de una región variable del brazo largo, que puede ir desde 15q11 a 15q14 y donde se han identificado 5 puntos de fractura recurrentes (BP1-BP5) (Distefano et al., 2019; Lusk et al., 2016). En este caso, el CEEA cumple con lo descrito, se observó en la forma de un cromosoma isodicéntrico con satélites en cada uno de los extremos (bisatelítico) (figura 1).
El cromosoma 15 es de gran interés por sus características especiales: 1- posee varios puntos con duplicaciones segmentales que lo hacen propenso a rearreglos, como translocaciones, inversiones, deleciones y duplicaciones y 2- posee regiones sujetas a impronta genómica (DiStefano et al., 2020; Makoff y Flomen, 2007). Todo ello lo vincula en gran proporción a condiciones patológicas involucradas principalmente con el neurodesarrollo (Tanet al., 2014; Fu et al., 2021).
De ahí, un diagnóstico probable para este probando sería el síndrome del cromosoma isodicéntrico del 15 (síndrome idic(15]), reconocido por sus manifestaciones clínicas, las cuales incluyen la hipotonía, el retraso del desarrollo motor, el retraso en el habla y ciertas características faciales dismórficas menores, que ocurren, por lo general, junto con DI y epilepsia. Aun así, muchas veces este síndrome no es fácil de identificar, debido a que los rasgos dismórficos no están presentes o son muy sutiles (por ejemplo, orejas grandes) y las malformaciones son raras, por tal motivo, el personal médico no solicita un cariotipo (Lusk et al., 2016). Adicionalmente, no se presentó edad materna avanzada al momento de la concepción, en ese momento era de 32 años, entonces, no se sospechó de un riesgo genético.
Las personas con una duplicación del cromosoma 15 exhiben un comportamiento característico que se describe como de espectro autista y en muchos casos también hay hiperactividad (Battaglia et al., 2010). El cuadro clínico esbozado en la literatura coincide con el fenotipo clínico de este probando. En lo común, los casos en mosaico manifiestan un fenotipo menos severo, aunque esto varía, dependiendo del porcentaje de células afectadas (Li et al., 2018). En este contexto, el cariotipo indicó un mayor porcentaje de células con presencia del marcador (74 %), en consecuencia, se esperan algunas o todas las manifestaciones clínicas del síndrome idic(15).
Mediante el ensayo de MLPA, se encontró que al menos los genes NDN (figura 2), MKRN3, MAGEL2 y SNRPN (figura 4) estaban duplicados. La región cromosómica involucrada (15q11-q13) está sujeta a impronta genómica, conocida como la región crítica del Síndrome de Prader Willi/Síndrome de Angelman (PWS/AS, del inglés Prader Willi Syndrome/Angelman Syndrome) y también incluye al gen UBE3A. Las duplicaciones aquí, principalmente de origen materno, se asocian con fenotipos que suponen hipotonía, trastornos del espectro autista, retraso del desarrollo, DI y convulsiones (Lusk et al., 2016; Sun et al., 2020). Se ha mencionado en la literatura que el gen UBE3A podría ser el principal responsable del síndrome idic(15), por cuanto la mayoría de los pacientes afectados comparten una duplicación materna – ya sea trisomía o tetrasomía- en relación con ese gen. De acuerdo con estos datos, la dosis génica de los genes expresados del alelo materno en dicha región es crítica para el adecuado desarrollo del cerebro (Battaglia et al., 2010).
Tanto la madre como el padre de este adolescente fueron evaluados a través de un cariotipo y una MLPA, dirigidos a descartar que el marcador fuera heredado. Cuando un marcador es heredado de un progenitor con un fenotipo normal, se considera que este no es el responsable del fenotipo observado en la descendencia (Milleret al., 2010; Watson et al., 2014). En este caso, el marcador ocurrió de novo, por lo cual, su presencia en mosaico podría ser la causa del cuadro clínico en el probando. El mecanismo propuesto para su formación es una no disyunción durante la meiosis de uno de los padres (presumiblemente en la oogénesis), seguido de un rescate de trisomía parcial (Kurtas et al., 2019; Sun et al., 2020).
En este trabajo no fue posible determinar el origen parental del cromosoma marcador, al respecto, se recomienda aplicar una PCR específica de metilación o la genotipificación de marcadores microsatélites. Así mismo, la técnica FISH podría contribuir a determinar los puntos de fractura del marcador con mayor resolución. Estos análisis adicionales permitirían confirmar el diagnóstico genético. La determinación de un marcador de novo también tiene implicaciones en cuanto al asesoramiento genético, particularmente, en la estimación del riesgo de recurrencia, que para los idic(15) no heredados se presume bajo (aunque se debe recomendar un diagnóstico preimplantacional en futuros embarazos, pues no se puede descartar mosaicismo germinal) (Lusk et al., 2016).
Finalmente, cabe destacar que, gracias al cariotipo se logró detectar que el material adicional en la región 15q no correspondía a una microduplicación, sino a un CEEA, al mismo tiempo, sin la MLPA no hubiera sido posible identificar con certeza la procedencia de ese CEEA, aspectos indispensables para el diagnóstico y para brindar asesoramiento genético acerca del riesgo de recurrencia.
5.3 Análisis del Caso 2
En el Caso 2, el gen RPH3AL estaba duplicado. Dado que no fue posible evaluar a los progenitores de este probando, fue imposible saber si dicho rearreglo es de novo o heredado y eso dificultó la determinación de su patogenicidad. Tampoco se tuvo la información sobre sus manifestaciones clínicas, lo cual impidió establecer una relación genotipo-fenotipo. Sobre este gen se sabe que está involucrado en los procesos de exocitosis tanto en células endocrinas como exocrinas y en el transporte de vesículas (Putchaet al., 2015; Varshney et al., 2021).
La duplicación de dicho gen ha sido reportada en personas con DI de novo y heredada de padres con fenotipo normal (Wellcome Trust Sanger Institute, s.f.), de tal modo, el desbalance detectado en la dosis génica del gen podría ser la causa de la DI del Caso 2. En la base de datos DECIPHER, una base de registro de desbalances cromosómicos y sus fenotipos, se considera esta duplicación como posiblemente patogénica (Wellcome Trust Sanger Institute, s.f.). Cuando es heredada de padres con fenotipo normal, es probable que las diferencias fenotípicas observadas se deban a que los puntos de fractura no sean idénticos por penetrancia incompleta o a que un alelo recesivo con una mutación patógena se exprese por la pérdida de su homólogo (Sarquis et al., 2011). Otras causas potenciales atañen a la interacción con otros genes, el bagaje genético de cada individuo, los factores epigenéticos, genéticos o no genéticos desconocidos (Costanzo et al., 2019; Ravnanet al., 2006). En esta situación particular, es necesario evaluar a los padres y conocer el fenotipo detallado para definir el posible origen de la DI.
6. Conclusiones
El elevado porcentaje de casos de DI sin diagnóstico de causalidad plantea el requerimiento de explorar nuevas técnicas de diagnóstico complementarias a las ya existentes. La técnica de MLPA es sensible y específica en la detección de aberraciones cromosómicas, las cuales son causas comunes de DI, como microdeleciones y/o microduplicaciones, así como marcadores supernumerarios.
El análisis citogenético, junto con la MLPA, es la mejor estrategia para el abordaje de personas con DI idiopática en países donde no se cuenta con la posibilidad de emplear la técnica de microarreglos, por su elevado costo o imposibilidades técnicas. Se recomienda establecer criterios de inclusión más estrictos para el estudio de pacientes, por ejemplo, el tener dos o más rasgos dismórficos, malformaciones congénitas o historia familiar de DI positiva; esto, con el objetivo de aumentar la probabilidad de detección de microdeleciones o microduplicaciones.
En definitiva, identificar una aberración cromosómica con potencial patogénico en pacientes con DI idiopática permitiría al médico tratante prever posibles complicaciones a futuro y evitaría someter al paciente a análisis adicionales innecesarios, dolorosos y/o con un costo económico alto para este o para el sistema de salud pública. Incluso, la detección de un CEEA generaría un alivio emocional en la familia de la persona afectada, al conocerse la probable causa de la DI.