








Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkActa Pediátrica Costarricense
versão impressa ISSN 1409-0090
Acta pediátr. costarric vol.21 no.1 San José Jan. 2009
Caso clínico
Enfermedad de Addison en una gemela de 19 meses
(Addison’s disease in a 19 months old twin girl)
Adriana Ulate-Campos,
Orlando Jaramillo-Lines
Servicio de Endocrinología, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera", Caja Costarricense de Seguro Social.
Correspondencia: Dr. Orlando Jaramillo Lines Servicio de Endocrinología, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera", correo electrónico: drojaramillo@yahoo.com
Resumen
La enfermedad de Addison, o insuficiencia adrenal primaria, es una entidad en la cual la corteza adrenal secreta cantidades insuficientes de glucocorticoides, mineralocorticoides y andrógenos debido a un daño adrenal.
Se presenta el caso de una niña de 19 meses con una historia de fatiga crónica, pérdida de peso, vómitos, hiporexia e hiperpigmentación en piel, labios y encías. Estos signos y síntomas tenían dos meses de evolución y empeoraron hasta que el diagnóstico correcto fue hecho y el tratamiento adecuado fue iniciado. Es una de las pacientes más jóvenes diagnosticada con enfermedad de Addison en Costa Rica, así como una edad de presentación temprana comparada con el promedio para esta patología. Se compara la paciente con su hermana gemela sana, de manera que los signos de la enfermedad son aún más evidentes y se presenta una revisión de la enfermedad de Addison como diagnóstico que debe estar presente en el diferencial de todo médico general.
Descriptores: Enfermedad de Addison, insuficiencia adrenal primaria, población pediátrica, hiperpigmentación, fatiga crónica.
Abstract:
Addison’s disease or primary adrenal insufficiency is a clinical entity in which the adrenal cortex secretes insufficient amounts of glucocorticoids, mineralocorticoids and androgens due to an adrenal noxa.
We report the case of a nineteen months old twin girl, who presented with a history of chronic fatigue, wasting, vomiting, hyporexia and hyperpigmentation in lips and gums. The symptoms continued for two months and worsened until the proper diagnosis was made and the treatment was established. This case represents one of the youngest patients diagnosed with Addison’s disease in our country, as well as an early age of presentation compared to the average age of diagnosis for this disease. We provide a comparison between the patient and her healthy twin sister, so the signs of the disease are even more evident.
Key words: Addison’s disease, primary adrenal insufficiency, children, hyperpigmentation, chronic fatigue.
La enfermedad de Addison (EA) fue descrita por primera vez por Tomás Addison hace 150 años como la asociación entre pérdida de peso, hiperpigmentación y destrucción de la glándula adrenal.(1,2) Ahora se conoce como una entidad en la cual la corteza adrenal es incapaz de secretar la cantidad necesaria de glucocorticoides, mineralocorticoides y andrógenos debido a un daño adrenal.(3,4) Por otro lado, la insuficiencia adrenal secundaria básicamente se presenta debido al déficit de hormona liberadora de corticotropina (CRH) y/o de hormona adrenocorticotrópica (ACTH) y existe, como consecuencia, una menor estimulación adrenal; sin embargo este tipo de insuficiencia adrenal no será el tema principal de este artículo.
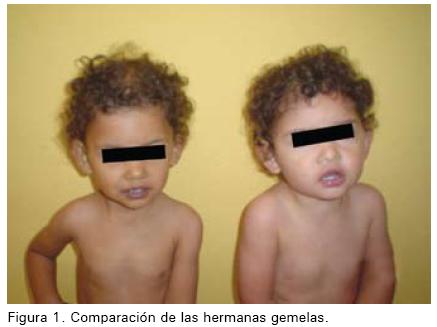
Se presenta el caso de una niña, el sexo es consistente con la epidemiología de la patología, pero ella tenía 19 meses cuando fue diagnosticada, convirtiéndola en uno de los casos de Addison más jóvenes diagnosticados en Costa Rica. Este caso resulta de particular interés porque la paciente tiene una hermana gemela quien estuvo sana en todo momento.
Caso clínico:
En el verano del 2008 una gemela de 19 meses, conocida sana, fue referida al servicio de Endocrinología del Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera (HNN) con una historia de fatiga crónica, náuseas, vómito, hiporexia y marcha atáxica. Dos meses previos a este evento los padres consultaron con un pediatra quien diagnosticó una neumonía y la niña recibió los antibióticos apropiados, con mejoría de los síntomas. Sin embargo los padres continuaron notando pérdida de peso en la niña, astenia y adinamia progresivas e hiperpigmentación; hallazgos que eran aún más evidentes al compararla con su gemela. Consultaron en varias ocasiones más sin recibir un diagnostico ni tratamiento certeros. Se debe mencionar que no existe historia familiar de consanguinidad ni de casos anteriores con síntomas similares, ni la niña ingirió ningún medicamento nuevo previo al inicio de los síntomas.
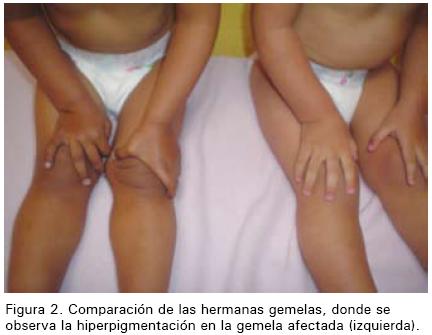
Al examen físico en el HNN se encontró una paciente letárgica, con un peso de 9,2 Kg (tercer percentilo para peso/edad) y una talla de 82 cm (50 percentilo talla/edad). Se notaron áreas de hiperpigmentación en labios, encías, areolas y línea media, y se evidenció hipotonía en las extremidades superiores e inferiores. Los signos vitales y el resto del examen físico fueron normales.
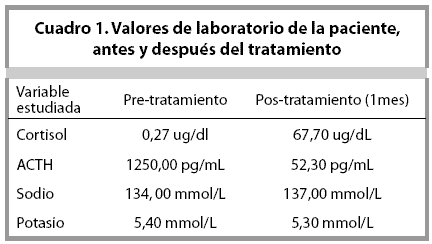
Estos hallazgos clínicos fueron consistentes con EA, y el diagnóstico fue confirmado por un cortisol sérico en 0, 27 ug/dl, y una ACTH en 1250,00 pg/ mL. Los electrolitos, sodio y potasio, se encontraron dentro de límites normales. Sus síntomas mejoraron un mes posterior al inicio del tratamiento con hidrocortisona y fluhidrocortisona, y la ACTH descendió a 52,30 pg/mL (Cuadro 1).
Discusión
La insuficiencia adrenal primaria tiene una prevalencia estimada de 90 a 140 por millón de personas.(1, 5) La EA es poco frecuente en la población pediátrica; es comúnmente diagnosticada entre la tercera y la quinta décadas de vida y es más habitual en mujeres. (3, 6)
En este caso los signos y síntomas de la enfermedad resultaron aún más llamativos para los padres, porque la diferencia se tornó más marcada y notoria con el paso del tiempo entre sus hijas. Un caso similar fue reportado en una gemela de 10 años en Indiana, cuatro años posteriores al diagnóstico la gemela idéntica no ha desarrollado la enfermedad. (7)
Addison describió los hallazgos de once casos, la mayoría de los cuales presentaban glándulas adrenales aumentadas de tamaño, hallazgo que él atribuyó a tuberculosis o a enfermedad maligna. También reportó un caso de un paciente con hiperpigmentación y vitíligo, sugiriendo un origen autoinmune.(2) Actualmente la frecuencia ha cambiado sustancialmente, las enfermedades autoinmunes se han convertido en la principal causa, cerca del 80% de los casos en adultos, y la tuberculosis ocupa el segundo lugar.(8,9) Estudios recientes demuestran que la incidencia de las enfermedades autoinmunes se ha incrementado en los últimos cincuenta años.(10) En los niños, del nacimiento a los dos años de vida la hiperplasia adrenal congénita es la principal causa de EA en ambos sexos, después de los dos años la adrenalitas autoinmune se convierte en la primera causa.(11)
Las etiologías de la EA se han dividido en tres grupos: 1) disgenesia adrenal, 2) destrucción adrenal y 3) esteroidogenesis inapropiada.(9,11) El primer grupo básicamente está compuesto por la hipoplasia adrenal congénita, mutaciones en los factores esteoidogénicos y una falta de respuesta a la ACTH. El segundo grupo está representado por el síndrome endocrino poliglandular, la adrenoleucodistrofia, la hemorragia adrenal y las infecciones. Una teoría para estos procesos autoinmunes es la selección negativa de los linfocitos en el timo; el riesgo para la autoinmunidad se encuentra en varios loci y probablemente se asocia con el complejo mayor de histocompatibilidad.(2, 12) Esta enfermedad también puede ser desencadenada por la combinación de anoxia y sepsis en el recién nacido.(13) El tercer grupo comprende la hiperplasia adrenal congénita y los desórdenes mitocondriales. La hiperplasia adrenal congénita, la principal causa de Addison en el periodo neonatal, tiene una incidencia de 1 en 15.000 nacidos vivos.(1, 14) La deficiencia enzimática más común es la de 21- hidroxilasa y los anticuerpos contra esta enzima son un marcador sensible y específico de autoinmunidad.(12) El tamizaje para esta patología ha disminuido la mortalidad significativamente. (1)
Los hallazgos clínicos de la EA son bastante sutiles y poco específicos al inicio por lo que con frecuencia no es diagnosticada de forma temprana, especialmente en la población pediátrica.(2) Esta patología debe ser considerada siempre, ya que puede ser letal cuando ocurre una crisis adrenal con colapso cardiovascular y no se instaura el tratamiento adecuado. La presentación se divide de acuerdo con la duración del cuadro clínico. La insuficiencia adrenal aguda se presenta con hipotensión, hipoglicemia, fatiga, debilidad, deshidratación y alteración del estado mental. En niños las convulsiones por hipoglicemia podrían ser la presentación inicial. (5) Mientras que la insuficiencia adrenal crónica se presenta con pérdida de peso, nauseas, vómitos, hiporexia, dolor abdominal recurrente, fuerza muscular disminuida, irritabilidad, hipoglicemia, hipotensión, "sed de sal", e hiperpigmentación.(4, 5, 9) La "sed de sal" y la hiperpigmentación son hallazgos clínicos que diferencian la insuficiencia adrenal primaria de la secundaria. (5) La hiperpigmentación es el resultado de la activación del eje hipotálamo-hipófisis con aumento de la liberación de la proopiomelanocortina y como consecuencia elevación en la concentración de hormona estimuladora de los melanocitos que produce hiperfunción de los melanocitos.(15) Este fenómeno típicamente se observa en las mucosas, pliegues de la piel, sitios de presión y cicatrices; principalmente en áreas expuestas al sol. La hiperpigmentación también se observa en efélides, las manchas café con leche de la neurofibromatosis y los léntigos.(16)
El abordaje clínico debe incluir siempre una historia clínica apropiada, tomando en cuenta aspectos como casos familiares similares, historia de consanguinidad, uso de medicamentos e infecciones recientes.(13) El diagnóstico se orienta por una concentración matutina (8am) baja de cortisol sérico (<150 nmol/L) y una concentración alta de ACTH (>20 pmol/L), estos resultados además ayudan a diferenciar entre una insuficiencia adrenal primaria y secundaria.(17,18) El diagnóstico también se puede realizar midiendo los niveles de cortisol antes y entre 30 y 60 minutos después de la aplicación de 250 μg de cosintropina (análogo sintético de ACTH). Un nivel de cortisol mayor a 500 nmol/L o un aumento del nivel de cortisol basal (>200 nmol/L) se consideran respuestas normales, mientras que un nivel menor a 500 nmol/ L se considera anormal o un resultado positivo. (17,19) La sensibilidad de este examen se relaciona con la severidad de la enfermedad; algunos pacientes con EA leve tienen un prueba de cosintropina normal por lo que requieren un seguimiento cercano.(17) Otros estudios sugieren que un prueba con una menor dosis de cosintropina (10 μg) es mαs sensible e intercambiable con el de la dosis estándar de 250μg, ya que este último resulta una estimulación suprafisiológica.(19) Algunos autores concluyen que ninguna de estas pruebas clasifican a los pacientes de manera adecuada, por lo que deben ser aplicadas con cautela y con una orientación clínica precisa.
Otros hallazgos de laboratorio en la insuficiencia adrenal primaria incluyen: hiponatremia, hipercalemia, hipoglicemia, creatinina sérica elevada, hipercalcemia, acidosis metabólica, aumento de la actividad de la renina, anemia, leucopenia y eosinofilia.(4,13)
La determinación de anticuerpos contra la glándula adrenal es útil para identificar, diagnosticar y tratar a los pacientes con EA de tipo autoinmune antes de que ocurra la destrucción completa de la glándula adrenal. (12, 18, 20) Un estudio demostró que en un plazo de 3 años el 50% de los pacientes con anticuerpos positivos contra la glándula adrenal desarrollaron algún grado de insuficiencia adrenal. (10) El hecho de que estos anticuerpos pueden estar ausentes en desordenes crónicos debe ser tomado en cuenta. (20)
En la EA las dosis de reemplazo de glucocorticoides son esenciales para garantizar la salud y la calidad de vida. La dosis recomendada de hidrocortisona oral es de 10-12 mg/m2/día, en niños esta cantidad se divide en tres tomas por día. Durante momentos de estrés la dosis de glucocorticoides debe ser duplicada y administrada de la forma usual, en tres tomas al día durante dos o tres días. (4, 11) En caso de una cirugía se recomienda administrar 25 mg/m2 I.V de hidrocortisona una hora previa a la cirugía y posterior a la misma se duplica la dosis diaria de mantenimiento administrada vía intravenosa cada seis horas. En caso de que se presente una situación de estrés aunada a intolerancia de la vía oral se debe brindar una dosis de hidrocortisona de 50-100 mg/m2 I.V según la condición del paciente. Las consecuencias de un reemplazo excesivo de glucocorticoides incluyen obesidad, hiperglicemia, diabetes mellitus, presión alta, pobre cicatrización y disminución de la velocidad de crecimiento, entre otros.(3,11) Cuando el paciente completa su crecimiento otra opción terapéutica es la administración de dexametasona en lugar de la hidrocortisona mencionada.
Esta patología también puede ameritar el reemplazo de mineralocorticoides, con fluhidrocortisona, a una dosis de 0,05-0,2 mg cada día.(11) Dosis elevadas de mineralocorticoides pueden resultar en efectos deletéreos en el sistema cardiovascular debido a hipertensión y niveles elevados de renina.(3,11)
Para monitorizar si el reemplazo hormonal es el apropiado parámetros como la pigmentación de la piel, la velocidad de crecimiento, la presión arterial y la ganancia de peso pueden ser útiles.(11) Se utilizan también parámetros de laboratorio tales como los niveles de ACTH, y electrolitos séricos.
El fallo en incrementar las dosis de glucocorticoides durante periodos de estrés constituye una causa de morbilidad en esta población. La mayoría de los estudios reportan tasas de mortalidad similares a las de la población general una vez que el diagnóstico se realiza y el adecuado reemplazo con hidrocortisona se inicia.(17) Sin embargo un estudio reciente en Suecia demostró una tasa de mortalidad dos veces mayor a la de la población general. El estudio atribuyó este hallazgo a un reemplazo inadecuado con glucocorticoides y a un incremento en la frecuencia de desordenes endocrinos asociados. Las principales causas de muerte fueron desordenes cardiovasculares seguidos por enfermedades malignas, endocrinológicas, respiratorias e infecciosas. (3)
La paciente presentó signos y síntomas de insuficiencia adrenal crónica porque, a pesar de la preocupación constante y permanente de los padres, existió una falta de sospecha clínica en los numerosos médicos a los que consultaron. Este caso resulta ejemplar debido a la comparación con la gemela sana; de modo que la EA debe ser considerada entre los diagnósticos diferenciales en un paciente que presente fatiga, pérdida de peso, náuseas, vômitos y sobre todo cuando se acompaña de hiperpigmentación.
Abreviaturas: EA, enfermedad de Addison; CRH, hormona liberadora de corticotropina; ACTH, hormona adrenocorticotrópica; HNN, Hospital Nacional de Niños "Dr. Carlos Sáenz Herrera".
Referencias
1. Shulman D, Palmert M, Kemp S. Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics 2007; 119:484-494. [ Links ]
2. Lovas K, Husebye E. Addison’s disease. Lancet 2005; 365:2058-2061. [ Links ]
3. Bergthorsdottir R, Leonsson-Zachrisson M, Odén A, et al. Premature mortality in patients with Addison’s disease: a population-based study. J Clin Endocrinol Metab 2006; 91: 4849–4853. [ Links ]
4. Gance-Cleveland B. Adaptation to Adisson’s disease in a child: a case study. J Pediatr Health Care 2003; 17:301- 310. [ Links ]
5. Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003. 361:1881–1893. [ Links ]
6. Barnard C, Kanani R, Friedman JN. Her tongue tipped us of. CMAJ 2004; 171:451. [ Links ]
7. Nebesio T, Ravi Shankar R. Hyperpigmentation in only one identical twin. J Pediatr Endocrinol Metab 2008; 21:7. [ Links ]
8. Simm PJ, McDonnell CM, Zacharin MR. Primary adrenal insufficiency in childhood and adolescence: Advances in diagnosis and management. J Paediatr Child Health 2004. 40:596-599. [ Links ]
9. Burk C, Ciocca G, Heath C et al. Addison’s disease, diffuse skin, and mucosal hyperpigmentation with subtle "Flu-like" symptoms-a report of two cases. Pediatr Dermatol 2008; 25:215-218. [ Links ]
10. Løvås K, Husebye ES. High prevalence and increasing incidence of Addison’s disease in western Norway. Clin Endocrinol (Oxf) 2002; 56:787-791. [ Links ]
11. Ten S, New M, MacLaren N. Clinical review 130: Addison’s disease 2001. J Clin Endocrinol Metab. 2001: 86:2909– 2922. [ Links ]
12. Barker J, Fain P, Eisenbarth G. Addison’s disease and type 1 diabetes. Curr Opin Endocrinol Diabetes 2005; 12:280-284. [ Links ]
13. Longui C. Insuficiencia adrenal primaria na infancia. Arq Brasileiros de Endocrinol Metabol 2004; 48:739-745. [ Links ]
14. Perry R, Kecha O, Paquette J, et al. Primary adrenal insufficiency in children: twenty years experience at the Sainte-Justine Hospital Montreal. J Clin Endocrinol Metab 2005; 90:3243-3250. [ Links ]
15. Vega J, Miranda A, Martínez G et al. Hyperpigmentation mimicking Laugier syndrome, levodopa therapy and Addison’s disease. J Eur Acad Dermatol Venereol 2003; 17:324-327. [ Links ]
16. Chhangani NP, Sharma P. Addison’s disease. Indian Pediatr 2003; 40:904-905. [ Links ]
17. Dorin RI, Qualls CR, Crapo LM. Diagnosis of adrenal insufficiency. Ann Intern Med 2003; 139:194-204. [ Links ]
18. Myhre A, Bjorses P, Dalen A, et al. Three sisters with Addison’s disease. J Clin Endocrinol Metab 1998; 83:4204- 4205. [ Links ]
19. González JG, De la Garza NE, Mancillas LG et al. A highsensitivity test in the assessment of adrenocortical insufficiency: 10 μg vs 250 μg cosyntropin dose assessment of adrenocortical insufficiency. J Endocrinol 1998; 159:275-280. [ Links ]
20. Laureti S, Casucci G, Santeusanio F, et al. X-linked adrenoleukidystrophy is a frequent cause of idiopathic Addison’s disease in young adult male. J Clin Endocrinol Metab 1996; 81:470-474. [ Links ]