










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina Legal de Costa Rica
versión On-line ISSN 2215-5287versión impresa ISSN 1409-0015
Med. leg. Costa Rica vol.27 no.1 Heredia mar. 2010
Revisión Bibliográfica
Embriología del desarrollo de los bronquios y el parénquima pulmonar
María José Acuña Navas, Enrique Arce Rodríguez, Ana María Baquero Barcenas, William Bonilla Mora, Karla Coto Chinchilla, Laura Guerrero Gamboa, Massiel Gutiérrez Porras, Jefry Jiménez Delgado, Carlos Leitón Villagra, José Pablo Madrigal Rojas, Carlos Monge Carvajal, Fernando Morales González, Natalia Núñez Delgado, Mónica Penón Portmann, Ana Gabriel Quirós Castro, Carolina Rivera Calderón *
Tutor : Dr. Maikel Vargas Sanabria+
* Estudiantes de Medicina, Departamento de Anatomía, Universidad de Costa Rica. (monicape11@gmail.com)
+ Médico Forense: Profesor Departamento de Anatomía.UCR. mvargassa@gmail.com
Resumen:
Las malformaciones congénitas del aparato respiratorio son un extenso número de patologías que oscilan en gravedad dependiendo del momento embriológico en que se dio el fallo y cuales mecanismos fueron afectados. En la revisión se profundiza en patologías congénitas como el secuestro broncopulmonar y la malformación adenomatoide quística.
Palabras clave:
Desarrollo pulmonar - Desarrollo traqueobronquial - Embriología pulmonar - Esbozo pulmonar - Malformaciones congénitas - secuestro broncopulmonar - Malformación adenomatoide quística - Enfisema Lobar Infantil.
Abstract:
Respiratory System Congenital malformations include a large number of disorders; their severity depends on the age of the egg and the systems which were involved. In this article, we review some congenital malformations such as bronchopulmonary sequestration and cystic adenomatoid malformation.
Key words:
Lung development – Tracheobronchial development - Pulmonary embryology - Tracheobronchial diverticulum - Pulmonary diverticulum - Congenital malformations - Bronchopulmonary sequestration - Cystic adenomatoid malformation - Child Lobar Emphysema.
Introducción
El árbol bronquial se origina desde la tráquea, la cual está formada de: epitelio respiratorio, músculo liso, tejido fibroso y anillos cartilaginosos incompletos hacia posterior.1 A nivel de la cuarta vértebra torácica está la carina, que es la bifurcación de la tráquea en los dos bronquios principales, uno relacionado con cada pulmón. El bronquio principal derecho es más corto, ancho y verticalizado que el izquierdo. Los bronquios principales se dividen dicotómicamente en bronquios lobares, los lobares se dividen y forman los bronquios segmentarios. Sucesivamente esto últimos se dividen en subsegmentarios grandes, luego subsegmentarios pequeños, bronquios terminales, hasta llegar a lo que se conoce como acino respiratorio, el cual se forma por el bronquiolo respiratorio, los sacos alveolares, y alveolos. En total tenemos aproximadamente 23 generaciones en el árbol bronquial hasta llegar a los alveolos.2 Luego de la primera generación, los bronquios están incluidos dentro del parénquima pulmonar.
Los pulmones son el órgano fundamental de la respiración. Estos son órganos pares, esponjosos, suaves, elásticos, y rosados. Se ubican dentro de la cavidad torácica, a ambos lados del mediastino, protegidos y aislados por la pleura.3 Cada uno tiene un ápex, que se ubica superior y una base relacionada con el diafragma. El hilio pulmonar se ubica en la cara mediastinal del pulmón, y en este se encuentran: el bronquio, la arteria y las venas pulmonares.3 En general, los pulmones abarcan desde C7 hasta T10. El pulmón derecho es más corto pero es más ancho y voluminoso que el izquierdo, este último se encuentra desplazado hacia lateral por el corazón. El pulmón derecho tiene tres lóbulos separados por dos cisuras (oblícua y horizontal); mientras que el izquierdo presenta dos lóbulos separados por la cisura interlobar.
Desarrollo de los pulmones
Durante las primeras etapas del desarrollo del sistema respiratorio presenta una amplia comunicación con el sistema digestivo. Al realizarse el plegamiento céfalo caudal y el plegamiento lateral del embrión, una porción de la cavidad del saco vitelino recubierta por endodermo quedará incorporada en el embrión para formar el intestino primitivo. 1
El intestino primitivo forma un tubo ciego a lo largo de todo el embrión, este tubo se encuentra entonces tanto en la región caudal como la región cefálica del embrión. Este tubo ciego de intestino primitivo está conformado por tres porciones según la región en la cual se ubique cada una, el intestino medio, intestino anterior cefálicamente e intestino posterior caudalmente. Sin embargo, con respecto a su desarrollo, el intestino primitivo se divide en cuatro secciones o segmentos en lugar de tres. Intestino faríngeo o faringe, el cual tiene especial importancia en el desarrollo de la cabeza y el cuello, un intestino anterior, que se encuentra en relación con el tubo faríngeo, además de un Intestino medio y un por último intestino posterior. 4
Con respecto al desarrollo pulmonar se tiene que en el embrión de aproximadamente 4 semanas, se forma una evaginación de la pared ventral del intestino anterior, la cual se denomina como divertículo respiratorio o esbozo pulmonar. Al mismo tiempo que se diferencia el aparato respiratorio se van constituyendo los vasos correspondientes para el sistema circulaciónventilación que se utiliza en la vida extrauterina. 4
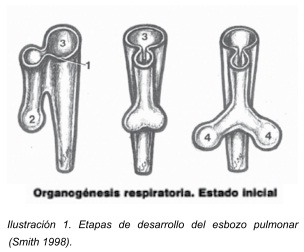
Desarrollo de traquea y bronquios.
Tempranamente el esbozo pulmonar se comunica de forma amplia con el intestino anterior, pero pierde esa comunicación directa conforme el esbozo pulmonar se extiende hacia caudal. A partir de ese momento, ambos elementos, el esbozo pulmonar e intestino anterior, quedarán separados por la aparición de dos rebordes longitudinales a los cuales se les denomina rebordes traqueoesofágicos. Después de su aparición, la fusión de estos rebordes, da lugar al tabique traqueo esofágico, con lo cual el intestino queda dividido en una porción dorsal que conforma el esófago, y en una parte ventral que forman la tráquea y los esbozos pulmonares. 5
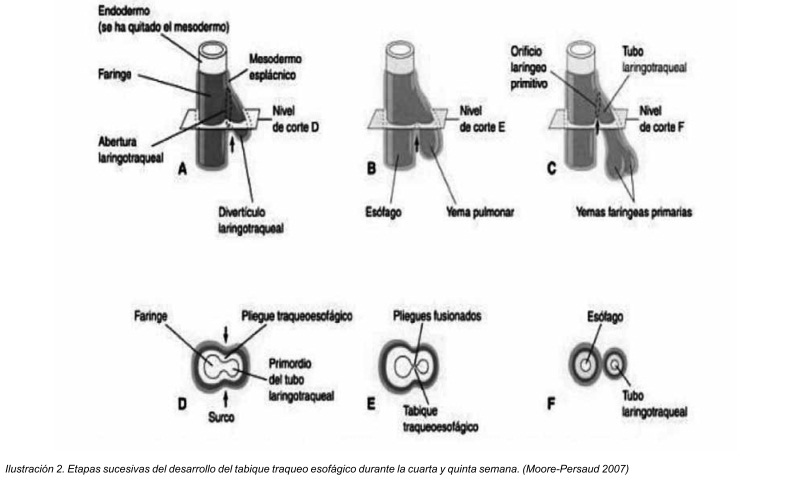
La tráquea es un dispositivo de sostén, su función es principalmente la de permitir un correcto paso de aire, a la vez de evitar que las compresiones que de órganos vecinos, la tráquea tiene un esqueleto formado a partir de mesodermo peri digestivo. El mesodermo esplácnico da lugar a los cartílagos traqueales, verdadero esqueleto traqueal, tiene una forma característica cilíndrica por su cara anterior y plana por su cara posterior.
El epitelio de revestimiento interno de la laringe, la tráquea los bronquios y pulmones es de tipo endodérmico. Los componentes cartilaginosos, musculares y conectivos de la tráquea y de los pulmones derivan del mesodermo esplácnico que circunda al intestino anterior del cual se derivan. Cuando se separan el intestino anterior de lo que ya es esbozo pulmonar, se forman la tráquea y las dos evaginaciones laterales que se conocen como esbozos pulmonares. Para la quinta semana ambos esbozos pulmonares se agrandan y forman los bronquios principales, izquierdo y derecho.6
Posteriormente el bronquio principal derecho se va a dividir en tres bronquios lobares (secundarios), mientras que el bronquio izquierdo se dividirá solamente en dos bronquios lobares. Lo cual insinúa la anatomía del adulto tal y como es, desde ese momento concuerda con la anatomía normal de los seres humanos, en los cuales finalmente se cuenta con la presencia de tres lóbulos pulmonares derecho y dos lóbulos pulmonares izquierdos. 5
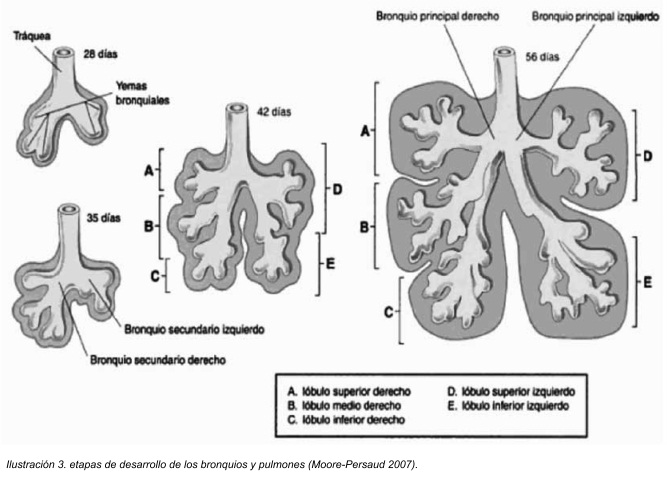
Al final de la quinta semana, los bronquios secundarios, se dividen en forma rápida y dicotómica hasta formar 10 bronquios segmentarios (terciarios) en el caso del pulmón derecho y 8 en el pulmón izquierdo, uno para cada segmento del pulmón. De esta manera se crean los segmentos broncopulmonares que se distinguen en el pulmón de un adulto. Al final del sexto mes se han originado aproximadamente 17 generaciones de subdivisiones. 4
Conforme ocurre el crecimiento y diferenciación de los esbozos pulmonares, en las estructuras ya citadas, estos se introducen en la cavidad corporal, específicamente en unos espacios destinados en el tórax para los pulmones, los cuales se conocen con el nombre de canales pericardio peritoneales. Estos canales se encuentran a cada lado del intestino anterior y son ocupados gradualmente por los esbozos pulmonares durante su desarrollo y aumento de tamaño. Los canales pericardio peritoneales, son separados de la cavidad peritoneal y pericárdica, por dos pliegues, los pliegues pleura peritoneal y pliegue pleuro pericárdico, los espacios resultantes de esta separación forman las cavidades pleurales primitivas. El mesodermo que reviste la parte externa del pulmón se desarrolla hasta convertirse en pleura visceral, mientras que la hoja somática de mesodermo que recubre la pared corporal del embrión interiormente, se transforma en pleura parietal. El espacio que quedará entre ambas pleuras es la cavidad pleural como tal.5
Las ramificaciones del árbol bronquial son reguladas por interacciones epitelio- mesenquimatosas de inducción molecular que veremos más adelante; que se dan entre el endodermo de los esbozos pulmonares y el mesodermo esplácnico que los rodea. Mientras se forman nuevas divisiones del árbol bronquial y este se desarrolla, los pulmones adoptan una posición cada vez más caudal de modo que al momento del nacimiento la bifurcación de la tráquea quede a nivel de la cuarta vértebra torácica.5
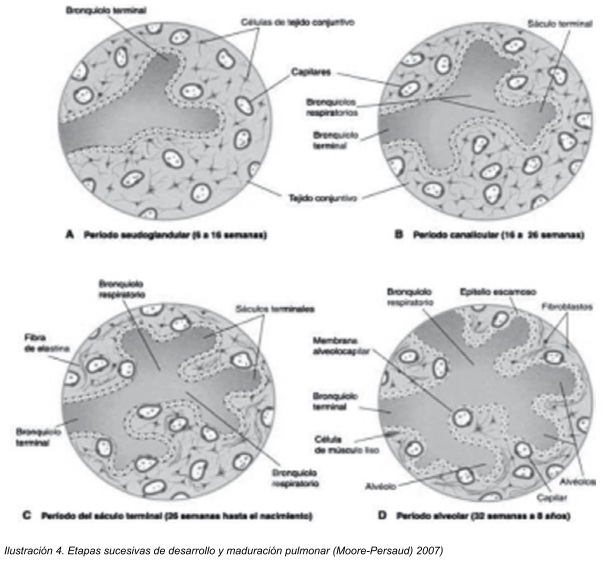
Mientras ocurren las divisiones de los bronquios primarios en secundarios, el mesénquima se agrupa alrededor de ellos constituyendo el esqueleto bronquial y la estructura morfológica, hemática, de sostén y relleno de cada uno de los lóbulos pulmonares, que al igual que los bronquios se forman tres lóbulos del lado derecho y dos del lado izquierdo. El mesénquima se condensa alrededor del brote endodérmico y da lugar a la musculatura de los bronquios, a los anillos cartilaginoso y al tejido conjuntivo, muscular y hemático. Entre cada uno de los lóbulos aparecen surcos de separación, quedando de esta forma independientes en su parte periférica, pero unidos por el hilio, estos surcos son las cisuras pulmonares. 6
Hasta el sétimo mes de desarrollo intrauterino los bronquiolos se dividen contínuamente en conductos cada vez más pequeños, este periodo se conoce como fase canalicular y abarca de la semana 16 a la 26 de la gestación, en esta fase la vascularización aumenta en forma constante y cada bronquiolo terminal se divide en dos o más bronquiolos respiratorios, los cuales a su vez se dividen en tres a seis conductos alveolares. 6
Posterior a ello, cuando algunas de las células cúbicas de los bronquiolos respiratorios se transforman en células delgadas y planas, conocidas como neumocitos tipo I, los cuales están íntimamente asociados a capilares tanto sanguíneos como linfáticos con los cual puede darse intercambio gaseoso y será posible la respiración para el niño. Al final del sexto mes aparecen también las células alveolares tipo II más conocidas como neumocitos tipo II, que son cúbicas en el adulto y son las células encargadas de producir factor surfactante, para disminuir la tensión superficial en la interfase sangre - alveolar.1
Mecanismos moleculares implicados en el desarrollo embriológico
Como se mencionó anteriormente, el pulmón se deriva de epitelio endodérmico, del cual se desprenden una gran cantidad de estirpes celulares: Células ciliadas, células basales, células claras, y células caliciformes en la porción proximal de las vías aéreas; neumocitos tipo I y II en las porciones distales de las vías. Mientras que el mesénquima que se deriva del mesodermo esplácnico origina al componente vascular, músculo liso, cartílago y demás tejido conectivo que se encuentra rodeando a los pulmones.7
El desarrollo del tracto respiratorio requiere de una serie de procesos bien orquestados que, cuando son alterados pueden producir rupturas en fases primordiales del desarrollo, especificación, crecimiento y diferenciación del tejido. El tema recurrente en estudios realizados mediante mutaciones génicas, es que el desarrollo pulmonar surge de una intrincada relación epitelio-mesénquima. Factores de crecimiento son los encargados de realizar la señalización e inducción de dicha interacción controlando así el destino celular, proliferación, migración y diferenciación.
Estudios realizados en ratones han demostrado que los progenitores de la tráquea y el pulmón son visualizados en su posición en el intestino anterior gracias a la expresión del gen Nkx2.1.8 Aunque el tiempo preciso en que dichas células son inducidas a crecer a partir del intestino anterior no está completamente dilucidado, investigaciones hechas a través de tejido endodérmico de ratones ha demostrado que las señales dadas por el factor de crecimiento fibroblástico (FGF) que proviene de las células cardiacas vecinas es primordial para la diferenciación del pulmón. In vitro, Foxa1 activa la transcripción del gen Nkx2.1 para definir así la diferenciación del linaje celular dentro del pulmón.9 En humanos, la pérdida de función por mutación en el gen Nkx2.1, localizado en el cromosoma 14q, ha sido descrito en infantes con fatiga respiratoria recurrente, además de disfunción de tiroides y de sistema nervioso central. 10
Formación del brote pulmonar
De la misma manera, en que las interacciones entre el epitelio y el mesénquima forman el esbozo traqueal esta misma interacción es la encargada de formar el brote pulmonar. Factores solubles han sido largamente implicados en la morfogénesis de la ramificación pulmonar, desde que fue descubierta que la transposición que realiza el mesénquima de la porción distal del pulmón en etapas tempranas, sobre el epitelio traqueal produce la ramificación supranumeraria así como la diferenciación de las células tipo II con los cuerpos lamelares y la expresión de la proteína C surfactante.11
Estudios en Drosophila han determinado que la división primaria de los pulmones es regulada espacialmente por un ligando del FGF: el gen Branchless, el cual activa a un receptor FGF (breathless) que a su vez estimula el desarrollo traqueal.12
El gen Branchless activa a un receptor FGF (breathless) en el endodermo que induce al desarrollo traqueal. Esta vía de señalización parece estar muy conservada evolutivamente en los mamíferos porque FGF10 y su receptor FGFR2b son críticos para la formación del pulmón. FGF10, localizado en zonas del mesénquima circundante alrededor de los brotes pulmonares distales, inducen y guían el crecimiento de los brotes, promoviendo la quimioatracción y la proliferación endodérmica.13 Varios estudios han correlacionado al ácido retinoico en la regulación de la expresión de FGF10 y en la formación de los brotes. Deficiencias de vitamina A o mutaciones en los receptores de dicha vitamina producen severas anormalidades en el pulmón, incluyendo fistulas traqueo esofágicas hipoplasias pulmonares, y agenesia del pulmón izquierdo.14
También la trascripción de los factores conocidos como T-box (Tbx) han sido ligados como reguladores de la expresión del FGF10. En embriones tempranos, Tbx y FGF10 son coexpresados en el mesodermo del intestino anterior.15 Así, la expresión de factores sobre el mesodermo del intestino anterior, incluidos entre ellos a los factores de crecimiento como el ácido retinoico, FGF10 y Tbx son críticos para la inducción primaria de los brotes pulmonares.
Tabicamiento Tráqueo-esofágico
La formación del septo tráqueo-esofágico se da junto con la formación del brote pulmonar primario. La expresión temprana de Nkx2.1 y Shh demarca el límite dorsoventral del endodermo del intestino anterior, distinguiéndose el primordio pulmonar del esófago. Esta separación dorsoventral está ausente o incompleta en embriones de ratones con mutaciones en Nkx2.1 y Shh, cuyo resultado es la aparición de fístulas tráqueo-esofágicas, así como hipoplasia de los pulmones.8
Asimetría Derecha Izquierda:
Tanto en humanos, como en otras razas de mamíferos, los pulmones tienen un patrón derecho-izquierdo asimétrico. Esta asimetría es dependiente de factores tempranos que determinan la especificación del eje derechaizquierda. Estos factores son regulados por los genes relacionados al Tgf-beta, como el receptor de Activina II, Lefty 1, Nodal y Pitx2.
Morfogénesis de la Ramificación:
La morfogénesis de la ramificación resulta en la formación de las vías aéreas de conducción hasta llegar a los bronquiolos, el cual es un proceso que involucra el crecimiento del esbozo pulmonar, elongación y división de las unidades terminales.15 El tamaño y forma del esbozo pulmonar está regulada, positiva y negativamente por señales emitidas por la interacción que se da entre el epitelio que crece y el mesénquima. Así como la inducción del esbozo pulmonar primario, la interacción entre FGF10-FGFR2B controla la formación del esbozo secundario. También FGF7 utiliza el mismo receptor, pero este regula la influencia de la ramificación pulmonar mediante la promoción de la proliferación y expansión de células epiteliales.16
Muchas vías de señalización regulan negativamente la proliferación del tejido epitelial del pulmón, contrarrestando así el efecto promotor de los factores de crecimiento fibroblástico sobre el esbozo pulmonar. Durante la elongación del esbozo pulmonar, FGF10-FGFR2B inducen a la expresión de Spry2, así como la expresión de BMP4. La familia SPRY son reguladores negativos de la señal de FGF, así como otras tirosínkinasas. Las reducciones en la actividad de Spry2 resultan en un aumento en la ramificación y proliferación celular. Inversamente, la sobreexpresión en dicho gen produce ramificación impar con un descenso en la proliferación de las células epiteliales.7 Así, la inhibición de SPRY2 sobre FGF10-FGFR2b, es parte esencial del control del tamaño del pulmón.
Pero la formación del esbozo pulmonar también está controlada por la expresión de SHH, el cual se expresa de manera importante en el epitelio del esbozo pulmonar distal, desde donde difunde para activar vías de señalización en el mesénquima adyacente a través de su receptor patched (Ptch1) y los factores de transcripción Gli.17
La familia del factor transformante beta (TGFb1) está también implicada en la morfogénesis de la ramificación, especialmente regulando la proliferación celular epitelial, y la deposición de matriz extracelular. Este factor es expresado en el mesénquima y se acumula alrededor de los ductos bronquiales y las ramas de las vías aéreas, donde el colágeno, fibronectina y proteoglicanos están presentes. Sobreexpresiones de Tfgβ1 tiene produce una señal regulatoria negativa sobre la morfogénesis de la ramificación, mientras que pérdidas de función de dicho factor, estimula a la ramificación pulmonar con una alta tasa de producción de células epiteliales.7
Establecimiento del patrón proximal-distal y la diferenciación celular:
Los diferentes linajes de las células epiteliales, están organizadas en diferentes patrones espaciales dentro de las vías aéreas, lo cual se vuelve morfológicamente evidente durante el período pseudoglandular. Células ciliadas, basales, secretorias, y neuroendocrinas son las células que constituyen el epitelio proximal, mientras que las células tipo I y II son las encargadas de crear el epitelio distal. La relación de linajes entre los diferentes tipos de células no han sido establecidas y la existencia de una célula progenitora, está aún bajo investigación.18
Experimentos de recombinación genética han demostrado que el fenotipo epitelial proximal, comparado con el distal, es dictado por factores solubles del mesénquima circundante, dentro de un período de tiempo restringido. El mesénquima pulmonar distal puede reprogramar el epitelio de la tráquea para crear células tipo II; mientras que, de manera inversa, el mesénquima de la tráquea puede inducir la diferenciación celular proximal, en el epitelio pulmonar distal. 19
Malformaciones congénitas
Las anomalías congénitas del aparato respiratorio, se originan debido a un problema en la expresión o inducción de tejidos explicadas anteriormente. Estas anomalías comprenden un extenso número de patologías que pueden comprometer el desarrollo de laringe, tráquea, bronquios, parénquima pulmonar, diafragma o pared torácica. Algunas se presentan como síndromes clínicos característicos, mientras otras sólo se consideran variaciones anatómicas que no requieren tratamiento. 20
Las malformaciones son a veces descubiertas en sonografías prenatales o a través de imagenología posnatal. Lesiones tales como malformación quística adenomatosa congénita (CCAM por sus siglas en inglés), secuestros, quistes broncogénicos y enfisema lobar congénito pueden ser asintomáticos en el nacimiento o en el momento en que se descubran más adelante en la vida. 21
Cada vez se hacen más comunes los diagnósticos de malformaciones congénitas de pulmón en adultos. Estas tienen manifestaciones únicas, a veces similares a otras patologías torácicas. La mayoría de estos pacientes han tenido resecciones parciales durante su infancia o han presentado infecciones recurrentes por años que son tratadas con medicamentos.
Ellos eventualmente manifiestan complicaciones que se pueden presentar agudamente y necesitar evaluación y tratamiento de emergencia.22 Se han descrito unos pocos casos en que la CCAM y el secuestro intralobar se han mantenido asintomáticos durante la vida, sin embargo, las complicaciones se desarrollan eventualmente prácticamente en todos los pacientes. La complicación más común es la pneumonía, que responde pobremente al tratamiento. Otras complicaciones pueden ser blastomas pleuropulmonares, malignificaciones (carcinomas) y pneumotórax o hemotorax. 21
Algunas malformaciones pueden permanecer asintomáticas o presentarse tardíamente en la vía como complicaciones por ejemplo infecciones. Por esta razón, y debido a la rareza de muchos de los desórdenes individualmente, no es posible saber su incidencia exacta. 23
Secuestro broncopulmonar:
Este puede ser definido como una porción del pulmón aislada de otra parte del pulmón de tal manera que no se encuentra con el árbol tráqueobronquial superior y recibe su irrigación sanguínea de una rama aberrante de la aorta y no de la arteria pulmonar. Esta lesión puede ser divida en 2 variantes, secuestro extralobar (ELS por sus siglas en inglés) y secuestro intralobar (ILS por sus siglas en inglés).24
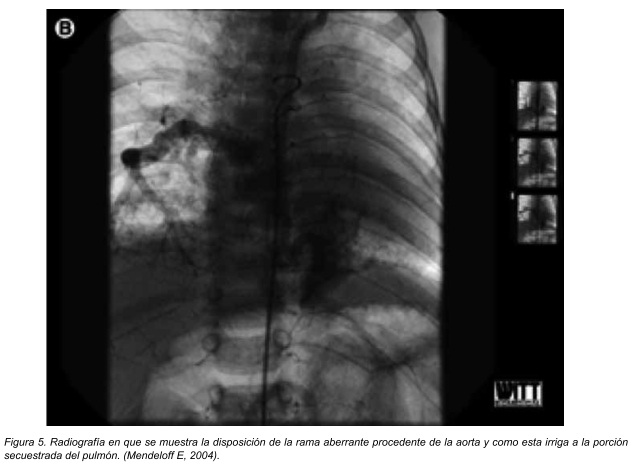
El ELS es una masa discreta del parénquima pulmonar que existe fuera del recubrimiento pleural del pulmón recibe irrigación sistémica y no está conectada con la vía aérea. Se ha postulado que esta se desarrolla cuando grupos de células independientes con potencial respiratorio proliferan del intestino primitivo caudal al primordio pulmonar que se desarrolla normalmente. Se ha demostrado que el gen homeobox Hoxb- 5 es necesario para el desarrollo de la ramificación normal de la vía aérea y se ha sugerido que estas anormalidades en el desarrollo observadas en el secuestro broncopulmonar se relaciona con una expresión anormal de los genes homebox. 25
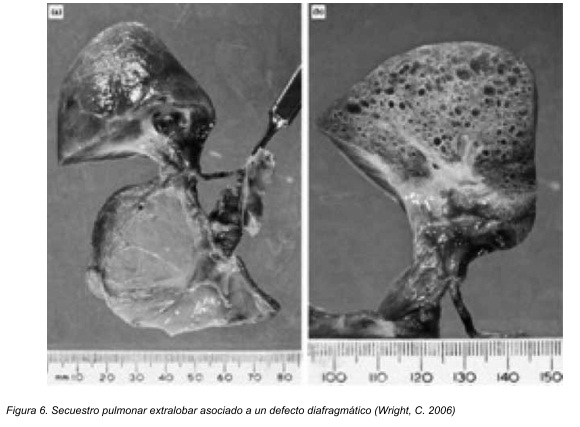
El ILS tiene características un poco diferentes. Esta malformación cuando está presente, se encuentra dentro de un lóbulo pulmonar y no tiene un recubrimiento pleural separado. Se localizan en el lóbulo inferior un 98% de las veces, 55% en el pulmón izquierdo y 45% en el derecho. Su irrigación está dada por una rama de la aorta torácica o de ramas provenientes de la aorta torácica y el tronco celiaco.24 Para este tipo de secuestro se ha propuesto una etiología adquirida con una lesión localizada del pulmón que probablemente concluye en obstrucción bronquial, lo que provocaría una cicatrización y fibrosis del parénquima relacionado. Esta inflamación recurrente puede estimular una respuesta neovascular dando como resultado el desarrollo de vasos colaterales en el ligamento pulmonar.26 Sin embargo también existen evidencias de que al menos algunas ILS son anormalidades en el desarrollo, que incluyen conexiones con el intestino anterior e histología similar a una malformación adenomatosa. 27
Enfisema Lobar Infantil
Se entiende por enfisema lobar congénito la hiperinsuflación de un lóbulo pulmonar. En este síndrome no hay destrucción del parénquima sino atrapamiento aéreo debido a un mecanismo valvular. Este mecanismo se produce por obstrucción parcial del bronquio ya sea extrínseca o intrínseca. La compresión extrínseca puede estar dada por alteraciones cardíacas o vasculares, adenopatías, etc. La compresión intrínseca puede deberse a alteraciones de la pared cartilaginosa bronquial o a la obstrucción del lumen por pliegues del epitelio, tapones mucosos, etc. EN el 50% de los casos no se ha identificado la causa de la obstrucción. 20
El aire atrapado que se encuentra distal a la obstrucción produce que los septos alveolares se rompan y se produzca una sobreexpansión del pulmón comprometido. Esta lesión raramente se relaciona con los lóbulos inferiores, y se encuentra en el lado derecho más frecuentemente que en el lado izquierdo.24
Malformación adenomatoide quística
La malformación adenomatoide quística del pulmón es una rara enfermedad (250 casos publicados).28 Se caracteriza por una proliferación anormal de elementos mesenquimales pulmonares. Sus rasgos comunes son: quistes recubiertos de epitelio columnar cuboideo tipo bronquial, comunicación con el árbol traqueobronquial, paredes constituidas por fibras elásticas y músculo liso, ausencia de glándulas mucosas y de cartílago.
La malformación adenomatoide quística del pulmón fue descrita por primera vez en 1949 y representa 25% de todas las malformaciones pulmonares congénitas. Su expresión clínica es variable, desde la muerte fetal o neonatal precoz por hipoplasia pulmonar o hidropesía no inmunitaria, hasta sobrevida por largos periodos asintomáticos seguidos de infecciones pulmonares recurrentes durante la niñez. El diagnóstico diferencial incluye el quiste broncógeno, el secuestro pulmonar y la hernia diafragmática.29
Su frecuencia como mencionamos antes es baja, más específicamente desde un caso por 5,000 nacimientos hasta uno por cada 20,000. Es una alteración pulmonar congénita poco frecuente (uno de cada 25,000 hasta uno de cada 35,000 nacimientos). Entre 80 a 95% de los casos son detectados en el periodo neonatal y aproximadamente 20% después del periodo neonatal. Se observa discreto predominio de las mujeres sobre los varones. 30
Se asocia a malformaciones del tipo: genitourinarias (agenesia renal y síndrome de Potter oligohidramnios, insuficiencia renal, facies característica, hipoplasia pulmonar), hidranencefalia, atresia yeyunal, hipoplasia pulmonar, secuestro extralobar, pectum excavatum, síndrome Prune-Belly (ausencia o hipoplasia congénita de la pared abdominal, severa dilatación no obstructiva del tracto urinario y criptorquidia bilateral), cardiopatías, así como transformaciones malignas (rabdomiosarcoma).31
Según el tamaño de los quistes y el aspecto microscópico, la malformación adenomatoide quística se ha clasificado en tres variedades: tipo I: grandes quistes; tipo II: múltiples quistes de tamaño inferior a 1 o 2 centímetros, y tipo III: masa homogénea y compacta con estructuras quísticas de tamaño inferior a 0,5 centímetros.32 La tipo III suele comprometer el lóbulo completo con estructuras que semejan bronquiolos entremezcladas con zonas recubiertas con epitelio similar a alveolos. Es poco frecuente (10%) y son las de peor pronóstico, cerca del 80% se presenta con polihidramnios. Recientemente Stocker agregó otros 2 tipos a las lesiones quísticas del aparato respiratorio nombrándolas tipo 0 y IV. La tipo 0 es incompatible con la vida y corresponde a un compromiso disgenético de todo el pulmón y la tipo IV corresponde a quistes de ubicación periférico. 20
En el período de recién nacido la MAQP se manifiesta como un síndrome de dificultad respiratoria. Al realizar una radiografía de tórax puede aparecer una masa sólida que luego tiende a airearse o desde el comienzo como un lóbulo con quistes de diferente tamaño en su interior. Estos quistes pueden insuflarse provocando un cuadro similar a un neumotórax hipertensivo con desplazamiento del mediastino. Frente a esta emergencia la punción de un quiste predominante puede dar tiempo para la lobectomía pulmonar de urgencia. Si el RN no nace sintomático y existía el antecedente de un eco cardiograma antenatal sospechoso, la radiología simple es insuficiente para el estudio del paciente, siendo necesario realizar una TAC de tórax. 20
El pronóstico y la sobrevida de los pacientes con esta afección es variable y depende del momento en que se hace el diagnóstico (antenatal o postnatal), la presencia o ausencia de hidrops o desviación de mediastino, la extensión (unilateral o bilateral), el tipo de quiste y el momento en que se decide la cirugía (antenatal in útero o postnatal). Son de alta letalidad las lesiones de tipo III, las bilaterales, el diagnóstico tardío y el hidrops fetal. 29
Esta malformación puede ser detectada en un ultrasonido antenatal de rutina, el cual es recomendado a las semanas 20-28 de gestación. Resonancia magnética fetal es un buen apoyo si es el estudio ultrasonográfico es de difícil interpretación. Aunque algunas lesiones pueden resolverse durante el embarazo y otros neonatos son asintomáticos al nacer, igual es importante recomendar a todos los casos sospechosos ser investigados más minuciosamente. La cirugía está recomendada para remover las lesiones después del nacimiento debido al alto riesgo de morbilidad por infección, pnemotórax y malignicidad. 33
Autores en la literatura mencionan que la etiología y la patogenia de la lesión son desconocidas, sin embargo en estudios recientes se ha experimentado con distintas moléculas de señalización y diferenciación embriológica para dar con la génesis del problema, y por ejemplo se ha encontrado el posible papel de el Factor de crecimiento fibroblástico (FGF10 por sus siglas en ingles) en la patogenia.
Dicha molécula (FGF10) es un factor de crecimiento mesenquimatosa, envuelto en interacciones epitelio-mesenquimatosas durante la morfogénesis de la ramificación pulmonar. Transferencia del gen FGF10 (sobreexpresión) en modelos de rata durante la fase pseudoglandular resulta en lesiones macrocísticas por clasificación ultrasonográficas confirmadas por evaluación con resonancia magnética en el día 7 posnatal. El mismo procedimiento en la fase canalicular tiene resultado parecido. Las manifestaciones clínicas de estas lesiones inducidas fueron sugestivas de malformación adenomatoide quística congénita humana. 34
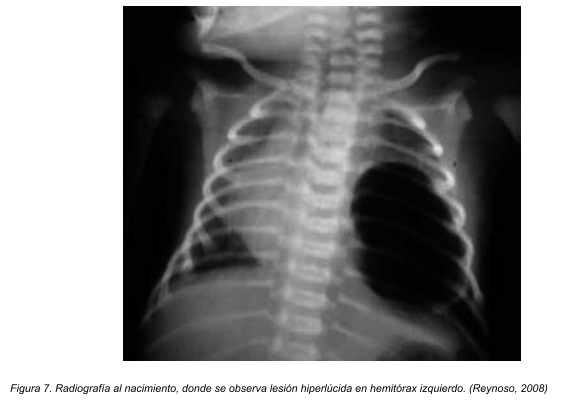
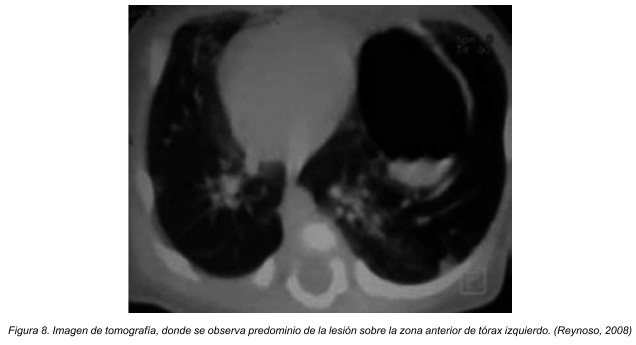
Vemos en las figuras 7 (radiografía de tórax) como se delimitan imágenes quísticas con nivel hidroaéreo y condensación periférica en hemitórax izquierdo. La tomografía axial computarizada torácica muestra una masa multiquística en base pulmonar izquierda con condensación parenquimatosa perilesional (Figura 8), imagenologia diagnóstica correspondiente a un paciente pediátrico masculino de 20 meses de edad.
Conclusiones
El adecuado desarrollo del sistema digestivo temprano, es de vital importancia para el embrión, ya que de esto depende también el desarrollo del sistema respiratorio del mismo.
El posicionamiento, diferenciación, desarrollo adecuado y el crecimiento del sistema respiratorio del feto, se ve influido y controlado de manera muy regulada por factores de transcripción.
El sistema bronquial se desarrolla en la quinta semana de vida fetal, por lo cual es una semana crítica y decisiva, si no se desarrolla este sistema, el embrión no podrá sobrevivir.
Las anomalías congénitas del aparato respiratorio comprenden un extenso número de patologías. Aunque la mayoría de veces su detección es temprana cada vez es más común su diagnóstico en adultos. El secuestro broncopulmonar es una malformación en el desarrollo del esbozo pulmonar en el intestino anterior, que de no ser tratada tempranamente puede derivar en el desarrollo de carcinomas u otras complicaciones. La etiología del enfisema lobar congénito no es muy clara, la obstrucción que la produce puede resultar en una forma localizada de hiperplasia pulmonar.
Resulta imprescindible para el ejercicio de la Medicina, tener conceptos embriológicos claros y poder correlacionarlos con la anatomía, ya que de esta manera y con el conocimiento de malformaciones, aún las que tienen una prevalencia baja, será posible en algunos casos aumentar la sobrevida de pacientes (usualmente pediátricos).
Recibido para publicación: 29 de octubre 2009 Aceptado: 15 de febrero 2010.
Referencias
1. Ross M. et al. (2006). Histología. Texto y Atlas Color con Biología Celular y Molecular. (4ª ed.). Buenos Aires, Argentina: Médica Panamericana. [ Links ]
2. Boron, W. y Boulpaep, E. (2005). Medical Physiology. (Updated Ed.). Pennsylvania, USA: Elsevier. P. 601. [ Links ]
3. Latarjet, M. y Ruiz, A. (2007). Anatomía Humana. Tomo 2. (4º ed.) Buenos Aires, Argentina: Médica Panamericana. P. 1148-1149. [ Links ]
4. Langman. (2006). Embriología médica con orientación clínica. (10º ed.). [ Links ]
5. Moore, K. (2007). Embriología clínica. Desarrollo del sistema respiratorio. (7º ed.). [ Links ]
6. Smith, V. (1998). Manual de embriología y anatomía general. [ Links ]
7. Have, T. (1991). Lung development in the mouse embryo. Exp Lung Res, 17, 111–130. [ Links ]
8. Minoo, P., Su, G., Drum, H., Bringas, P. y Kimura, S. (1999). Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(-/-) mouse embryos. Dev Biol, 209, 60–71. [ Links ]
9. Serls, A. E., Doherty, S., Parvatiyar, P., Wells, J. M., (2005). Deutsch GH. Different thresholds of fibroblast growth factors pattern the ventral foregut into liver and lung. Development, 132, 35–47. [ Links ]
10. Krude, H., Schutz, B. y Biebermann, H. et al. (2002). Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2–1 haploinsufficiency. J Clin Invest, 109, 475–480. [ Links ]
11. Shannon, J. M. e Hyatt, B. A. (2004). Epithelialmesenchymal interactions in the developing lung. Annu Rev Physiol, 66, 625–645. [ Links ]
12. Sutherland, D., Samakovlis, C. y Krasnow, M. A. (1996). Branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell, 87,1091–1101. [ Links ]
13. Weaver, M., Dunn, N. R., Hogan, B. L. (2000). Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development, 127, 2695–2704. [ Links ]
14. Desai, T. J., Chen, F., Lu, J., et al. (2006). Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Dev Biol., 291, 12–24. [ Links ]
15. Hogan, B. L. (1999). Morphogenesis. Cell, 96, 225–233. [ Links ]
16. Nogawa, H. e Ito, T. (1995). Branching morphogenesis of embryonic mouse lung epithelium in mesenchyme-free culture. Development, 121:1015–1022. [ Links ]
17. Bellusci, S., Furuta, Y., Rush, M. G., Henderson, R., Winnier, G. y Hogan, B. L. (1997). Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development, 124, 53–63. [ Links ]
18. Rawlins, E. L., Hogan, B. L. (2006). Epithelial stem cells of the lung: privileged few or opportunities for many? Development, 133, 2455–2465. [ Links ]
19. Shannon, J. M., Nielsen, L. D., Gebb, S. A. y Randell, S. H. (1998). Mesenchyme specifies epithelial differentiation in reciprocal recombinants of embryonic lung and trachea. Dev Dyn, 212, 482–494. [ Links ]
20. Aldunate, M. (2001). Malformaciones pulmonares congénitas. Rev Chil Pediatr, 72, 52-7. [ Links ]
21. Laberge, J. (2005). Asymptomatic congenital lung malformations. Seminars in Pediatric Surgery. 14, 16-33 [ Links ]
22. Shanmugam, G. (2005). Adult Congenital Lung Malformations. European Journal of Cardio-thoracic Surgery, 28, 483-489. [ Links ]
23. Wrigth, C. (2006). Congenital Malformations of the Lung. Current Diagnostic Pathology, 12, 191-201. [ Links ]
24. Mendeloff, E. (2004). Sequestrations, Congenital Cystic Adenomatoid Malformations, and Congenital Lobar Emphysema. Semin Thorac Cardiovasc Surg, 16, 209-214. [ Links ]
25. Volpe, M., Archivachotikul, K. y Bahn, I. et al. (2000). Association of bronchopulmonary Sequestration with expression of the homeobox protein Hox-%. J Piediatr Surg, 35, 1817-1819. [ Links ]
26. Bratu, I., Fleageole, H., Chen, M. F., et al. (2001). The multiple facets of pulmonary sequestration. J. Pediatr Surg, 36, 784-790 [ Links ]
27. Imai, Y. y Mark, E. (2002). Cystic adenomatoid Change is Common to Various forms of Cystic Lung Disease in Children. Arch Pathol Lab Med, 126, 934-940. [ Links ]
28. Macdonald, M. R., Vito, F., Cutz, E. y Crysdale, W. S. (1996). Congenital Cystic Adenomatoid Malformation of the Lung Referred as "Airway Foreing Body". Arch Otolaryngol Head Neck Surg, 122, 333-337. [ Links ]
29. Rossel, K., Salinas, R., Kakarieka, E. y Espinosa A. (1996). Malformación adenomatoidea quística pulmonar del recién nacido. Rev Chil Pediatr, 67, l67-17l. [ Links ]
30. Mondragón, J., et al. (2004). Malformación adenomatoidea quística pulmonar. Informe de un caso de presentación tardía. Acta Pediatr Mex, 25, 333-336. [ Links ]
31. Sánchez, J. R. (2005). Malformación adenomatoidea quística congénita del pulmón. Rev Arch Med Camagüey, 9, 51-58. [ Links ]
32. Stocker, J. T., Madewe!, J. y Drake, R. (1977). Congenital cystic adenomatoid malformation of the lung. Clasification and morphologic spectrum. Hum Pathol, 8, 155-171. [ Links ]
33. Xue-lian, LI., et al. (2007, Dec.). Antenatal Diagnosis and Outcome of 12 Congenital Cystic Adenomatoid Malformation of Lung, 18, 4, 289-295. [ Links ]
34. Gonzaga, S., et al. (2008). Cystic adenomatoid malformations are induced by localized FGF10 overexpression in fetal rat lung. Am J Respir Cell Mol Biol, 39, 3, 346-55. [ Links ]
35. Reynoso, E., et al. (2008). Malformación adenomatoide quística en un recién nacido. Reporte de un caso y revisión de la literatura. Rev Med Hosp Gen Mex, 71, 1, 36-41 [ Links ]

