











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las toxinas paralizantes (PSP: Paralytic Shellfish Poisoning) son producidas a partir de los dinoflagelados de los géneros Alexandrium, Gymnodinium y Pyrodinium (3)-(5). Estos organismos son consumidos por los moluscos bivalvos como parte de su dieta natural. Si el dinoflagelado contiene las toxinas, estas son acumuladas activamente por los moluscos concentrándose en su hepatopáncreas (6). Las toxinas PSP se pueden clasificar por su estructura en tres subgrupos; el grupo N-sulfocarbamoil: B1, B2, C1, C2, C3, C4; el grupo decarbamoil: dcSTX, dcNeo, dcGTX1, dcGTX2, dcGTX3 y dcGTX4 y el grupo carbamoil: STX, Neo, GTX1, GTX2, GTX3, GTX4 (4)-(7). Estas toxinas afectan el sistema nervioso central ya que se unen reversiblemente con alta afinidad a los sitios de unión en el canal de sodio; de esta manera bloquean el paso de los iones de sodio hacia el interior de la célula, inhibiendo el desarrollo del potencial de acción y la generación del impulso nervioso, provocando parálisis cardiorrespiratoria que puede llevar a la muerte del individuo (8)-(10).
En Costa Rica la obtención de los moluscos bivalvos se lleva a cabo por medio de dos metodologías: ostricultura o cultivo de ostras y extracción del producto del medio natural. En la actualidad, el cultivo de moluscos bivalvos se desarrolla activamente en el Golfo de Nicoya, donde existen cuatro granjas ostrícolas que producen la ostra japonesa (Crassostrea gigas) (11) para consumo local, con expectativas de exportar a corto plazo. La extracción es una práctica que se ha realizado a lo largo del litoral Pacífico costarricense, donde se puede mencionar especies de interés como la piangua (Anadara tuberculosa), la almeja arenera (Chione fluctifraga), la concha perla u ostra perlera (Pteria sterna); la barba de hacha o concha abanico (Pinna rugosa) y el ostión vaca (Spondylus calcifer) (11).
En el 2000, se presentó en Costa Rica un evento importante de Floraciones Algales Nocivas (FANs), para el cual se registraron 77 casos de intoxicación en humanos y la muerte de animales (cerdos y perros) por el consumo de moluscos bivalvos contaminados con toxinas PSP generadas por dinoflagelados. Por lo anterior, en el 2001, se publicaron los Decretos Ejecutivos 29328-MAG-S y N° 39669-MAG-S, donde se crea la Comisión Interinstitucional para la Prevención y Control de la Marea Roja en Costa Rica (11) y dicha Comisión se convierte en una comisión asesora del Laboratorio Nacional de Servicios Veterinarios (LANASAVE), perteneciente al Servicio Nacional de Salud Animal (SENASA), quien decretará las vedas y tomará las acciones sanitarias correspondientes en materia de marea roja tóxica (12). A causa de este evento registrado en el 2000, se estableció una prohibición total a la extracción y comercialización de moluscos bivalvos mediante el Decreto Ejecutivo Nº 29184-S-MAG37. La concha perla, la concha abanico y el ostión vaca permanecen vedados desde entonces.
El LANASEVE es el ente que realiza desde el 2000 la determinación de las toxinas PSP en los moluscos bivalvos, como parte del monitoreo nacional organizado por el SENASA, el Instituto Costarricense de Pesca y Acuicultura (INCOPESCA) y la Universidad Nacional (UNA). La determinación se lleva a cabo por medio del método de bioensayo en ratón, de acuerdo con el método AOAC 959.08 (13), el cual es reconocido por la Comisión de Regulación de la Unión Europea en su reglamento (CE) N° 2074/2005 como método de referencia para la determinación de las toxinas paralizantes (PSP) (4), (14); y por el Codex Alimentarius como método para control de rutina (15). No obstante para lo anterior, la Unión Europea en el mismo reglamento, indica que las pruebas biológicas deben ser reemplazadas tan pronto sea posible, por un método validado internacionalmente (4).
La principal desventaja del método de bioensayo en ratón es que no expresan de forma separada la concentración de cada análogo de la saxitoxina, reportando todo en términos de saxitoxina total, por lo que no es posible identificar el tipo de toxina presente. Para el LANASEVE surge la necesidad de implementar una metodología analítica por medio de la cual se obtenga el perfil de toxinas para cada ítem de ensayo, y así cumplir con los requisitos establecidos por las normativas de referencia y el mercado internacional (16)-(18).
Por lo anterior, y en respuesta a las necesidades de la institución SENASA relacionadas con el compromiso país con las exigencias del mercado internacional, propone el objetivo de llevar a cabo la validación de una técnica para la determinación de las toxinas PSP en moluscos bivalvos mediante cromatografía líquida acoplada a reactor post-columna con detector de fluorescencia. Los beneficios esperados para la institución son la implementación del ensayo en las diferentes matrices, así como el establecimiento de tendencias que permitan la toma de acciones sanitarias para la protección de la salud pública.
Materiales y métodos
Las toxinas paralizantes evaluadas fueron las disponibles en el periodo de estudio como materiales de referencia del National Research Council of Canada, único proveedor a nivel mundial, a saber; STX, dcSTX, NEO, GTX1, GTX2, GTX3, GTX4, GTX5, dcGTX2, dcGTX3, C1 y C2.
Método de extracción
Para la extracción de las toxinas PSP se evaluó la metodología descrita por Rourke et al. (1), quienes utilizaron el ácido tricloroacético al 30% (m/v) en una etapa de purificación para la eliminación de las proteínas.
Para evaluar el desempeño de la metodología se inocularon muestras de ostra japonesa con PSP a un nivel de concentración intermedio de la curva de calibración de cada toxina. Se utilizó el valor medio del porcentaje de recuperación obtenido, como criterio de evaluación de la veracidad y se comparó con los criterios de aceptación definidos por Codex Alimentarius STAN 292 (19).
De las muestras de moluscos bivalvos se midió una masa de 100 g de músculo homogenizado, a los cuales se les agregó 100 mL de HCl 0,1 mol/L. Se verificó que el pH se encontrara entre 2 y 4, para posteriormente calentar el homogenizado hasta ebullición por 5 min, dejando luego enfriar a temperatura ambiente. El volumen total se ajustó a 200 mL con HCl 0,1 mol/L, se centrifugó por 10 min a 4000 s-1; del sobrenadante obtenido se colocaron 500 µL en un tubo de centrífuga de 2 mL, se agregaron 25 µL de ácido tricloroacético al 30% m/v, mezclando el contenido del tubo utilizando un vortex. Este se centrifugó a 4000 s-1 por 5 min, luego se agregaron 20 µL de NaOH 1,0 mol/L, se centrifugó a 4000 s-1 por 5 min, para finalmente filtrar el sobrenadante con un acrodisco de 0,22 µm. El filtrado se recolectó en un vial para automuestreador.
Método cromatográfico
Para la implementación del método cromatográfico se evaluaron y optimizaron las condiciones establecidas por Franco y Fernández (2), cuyo procedimiento consiste en realizar tres análisis cromatográficos para la determinación de los grupos de las toxinas, todos de modo isocrático.
En la separación de las toxinas NeoSTX, dcSTX, STX, se utilizó como fase móvil 95% de octanosulfonato de sodio 0,5 mmol/L, ácido fosfórico 10 mmol/L a pH 7,2 y 5% de acetonitrilo. La fase móvil utilizada en la separación de las toxinas GTXs y dcGTXs fue octanosulfonato de sodio 1,5 mmol/L, ácido fosfórico 10 mmol/L a pH 7,0. Las toxinas C1 y C2 se separaron con un 95% de fosfato de tetrabutilamonio 2,0 mmol/L a pH 6,5 y 5% de acetonitrilo.
En todos los casos, se realizó la derivatización postcolumna, empleando como reactivos derivatizantes una disolución de ácido peryódico 7 mmol/L, ácido fosfórico 50 mmol/L a pH 9 (reactor 1) y al ácido acético 0,5 mol/L (reactor 2) (2).
Se evaluó también la metodología planteada por Rourke et al (1), quienes establecen un programa en gradiente para la separación de las toxinas NeoSTX, dcSTX, STX, GTXs y dcGTXs, empleando como fase móvil: A: ácido heptanosulfónico 11 mmol/L, ácido fosfórico 5,5 mmol/L (pH 7,1); B: 88,5% ácido heptanosulfónico 11 mmol/L, ácido fosfórico 16,5 mmol/L, 11,5% acetonitrilo (pH 7,1). La separación de las toxinas C1 y C2 utilizó como fase móvil fosfato de tetrabutilamonio 2,0 mmol/L (pH 5,8) de modo isocrático. En ambos casos, se efectuó la derivatización postcolumna con los siguientes reactivos: ácido peryódico 5 mmol/L, ácido fosfórico 100 mmol/L (pH 7,8) (reactor 1) y ácido nítrico 0,75 mol/L (reactor 2).
Para la determinación de los grupos STXs y GTXs se utilizó una columna Zorbax Bonus - RP C14, 150 x 4.6 mm d.i., 3.5 µm de tamaño de partícula, con reactor post-columna (pickering), un flujo de fase móvil de 0.8 mL/min, un flujo de oxidante y ácido de 0.4 mL/min, temperatura del reactor de 85°C, temperatura del horno de columna de 40°C, un volumen de inyección de 20 µL para GTXs y STXs, el tiempo de corrida fue de 28 min. El gradiente se programó a 100% de Fase A los primeros 8 minutos, posteriormente 100% de Fase B del minuto 8 al minuto 16, y finalmente 100% Fase A del minuto 16 al minuto 28.
Las condiciones cromatográficas para la determinación de las toxinas Cs fueron una columna Zorbax Eclipse XDB-C8, 150 x 4.6 mm d.i., 5 m de tamaño de partícula, con reactor post- columna (pickering), un flujo de fase móvil de 0.8 mL/min, un flujo de oxidante y ácido de 0.4 mL/min, temperatura del reactor de 85°C, temperatura del horno de columna de 40°C, un volumen de inyección de 20 µL para Cs, el tiempo de corrida fue de 24 min.
Validación del método
Para evaluar las características de desempeño según lo establecido en la normativa vigente (19-20), se realizó la validación del método a partir de la determinación de los siguientes parámetros:
Linealidad y rango de trabajo
Se prepararon cuatro curvas de calibración para cada toxina, cada una con cinco disoluciones patrón. Los patrones se analizaron en el HPLC/FLD acoplado a reactor post columna, a partir de los datos de señal y concentración se graficaron las curvas de calibración; con el coeficiente de correlación y determinación se evaluó la linealidad. También se evaluó la similitud de las curvas preparadas para cada toxina mediante las pruebas de Tuckey y Fisher del análisis ANOVA, a un 95% de confianza. El rango de trabajo fue definido de acuerdo con la linealidad y el límite máximo permitido.
Sensibilidad
La sensibilidad de calibración se define como el cambio de la respuesta obtenida dividido por el correspondiente cambio de la concentración del analito; ésta se determinó como la pendiente de la ecuación de la curva de calibración promedio de cada toxina.
Veracidad
La veracidad se evaluó a través del porcentaje de recuperación. Se realizaron seis repeticiones a un nivel de concentración, para las toxinas STXs y GTXs, y dos repeticiones a un nivel de concentración para las toxinas Cs. Se tomó como base el rango lineal de trabajo de cada analito para establecer el nivel de concentración, en todos los casos se empleó como matriz muestras de ostra japonesa.
Precisión
La repetibilidad instrumental se determinó analizando la dispersión de seis lecturas diferentes de un punto de la curva (el tercer patrón de la curva de calibración de cada toxina) y se expresó como coeficiente de variación.
La repetibilidad de la metodología se estableció mediante la evaluación de la dispersión de seis réplicas de una muestra de concentración conocida para el caso de las GTXs y STXs y dos réplicas para las toxinas Cs, analizadas bajo las mismas condiciones. Se tomó como base el rango lineal de trabajo de cada analito para establecer el nivel de concentración. En todos los casos se empleó como matriz muestras de ostra japonesa, se determinó la desviación estándar y se calculó el coeficiente de variación correspondiente.
Límite de detección
Se realizó la lectura independiente de 11 blancos matriz para el caso de STXs y GTXs y de 20 blancos matriz para las toxinas Cs. Se determinó el promedio de las señales obtenidas de los blancos matriz y se le sumó tres veces el valor de la desviación estándar obtenida del conjunto de datos, dicho valor se interpoló en la curva de calibración promedio, para obtener la mínima concentración detectable.
Límite de cuantificación
A partir de la lectura de los blancos matriz empleados para la determinación del límite de detección, se determinó el promedio de la señal del equipo obtenida y su desviación estándar. El valor obtenido de la suma del promedio más 10 veces la desviación estándar se interpoló en la curva de calibración promedio, para obtener la concentración mínima cuantificable. Se fortificaron tres muestras cerca del límite de cuantificación de cada toxina y se determinó el porcentaje de recuperación para verificar la veracidad del método en el límite de cuantificación.
Resultados y discusión
Rendimiento de recuperación del método de extracción
El cuadro 1 resume los valores medios de los porcentajes de recuperación obtenidos con sus respectivas desviaciones estándar que describen el comportamiento de la precisión expresada. Los porcentajes de recuperación obtenidos para las toxinas del grupo STXs, GTXs y Cs, al aplicar el método de Rourke et al. (1), son aceptables según los criterios de veracidad establecidos por CODEX STAN 292-2008, donde se indica que porcentajes de recuperación entre 50 y 130% son aceptables para este tipo de matriz (19). El ácido tricloroacético empleado en la etapa de extracción propuesta por Rourke et al. (1), permitió precipitar la proteínas presentes en la muestra y de esta manera eliminar cualquier interferencia que pudiera influir sobre las propiedades analíticas de los analitos reduciendo así el efecto matriz.
Condiciones cromatográficas
Se utilizaron dos referencias para el establecimiento de las condiciones cromatográficas para los distintos subgrupos de toxinas PSP. En las figuras de 1 a 6 se presentan los resultados de esta optimización.
Cuadro 1 Valores promedio de porcentajes de recuperación obtenidos de toxinas de los grupos STXs, GTXs y Cs.
| Neo | dcSTX | STX | GTX4 | GTX1 | dcGTX3 | |
| % Rec | 84 | 114 | 111 | 66 | 81 | 90 |
| Desv Est | 4,8 | 3,2 | 3,4 | 10,2 | 5,7 | 3,3 |
| GTX5 | dcGTX2 | GTX3 | GTX2 | C1 | C2 | |
| % Rec | 77 | 96 | 102 | 115 | 72 | 58 |
| Desv Est | 6,3 | 3,0 | 2,7 | 4,6 | 3,2 | 0,9 |
En las figuras 1 y 2 se muestran los cromatagramas obtenidos al utilizar las dos metodologías propuestas para la separación de las STXs. Al aplicar lo establecido por Franco et al. (2) (figura 1), no se observa la separación de todas las STXs, lo que implica una posible coelución en dos picos cromatográficos. En la figura 2 se muestra la separación de las STXs al utilizar el método propuesto por Rourke et al. (1), donde se utiliza la fase móvil con gradiente. Con esta metodología, se logra la separación satisfactoria de las toxinas STXs.
Las figuras 3 y 4 muestran los cromatogramas obtenidos para las toxinas del grupo GTXs. Utilizando la metodología establecida por Franco et al. (2), como se obseva en la figura 3, no se logra la separación adecuada de este grupo de toxinas. La dcGTX3 y la GTX5 se obtienen en el mismo pico cromatográfico, lo cual se puede explicar por la ausencia del acetonitrilo en la fase móvil. La figura 4 muestra el cromatograma obtenido al aplicar el método de Rourke et al. (1); los compuestos se retienen en la columna por interacciones hidrofóbicas, en este caso, el acetonitrilo disminuye la polaridad de la fase móvil, el efecto hidrofóbico disminuye y se logra la elución de los compuestos.
Las figuras 5 y 6 muestran los cromatogramas obtenidos para las toxinas C1 y C2 al aplicar las condiciones cromatográficas establecidas por Franco et al. (2) y Rourke et al. (1) respectivamente. En la figura 5 se observa como al aplicar el método propuesto por Franco et al. (2) no se obtiene una separación satisfactoria de las toxinas ensayadas, sin embargo, al eliminar el 5 % de fase orgánica y disminuir el pH a 5.8, según lo establece Rourke et al. (1) (figura 6), se logra la resolución adecuada de los picos, siendo, por tanto, el método aceptable para la separación de este grupo de toxinas.
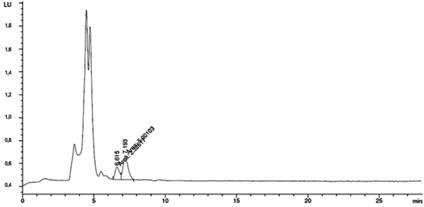
Figura 1 Cromatograma obtenido para las toxinas STXs al aplicar las condiciones cromatográficas establecidas por Franco et al. (2).
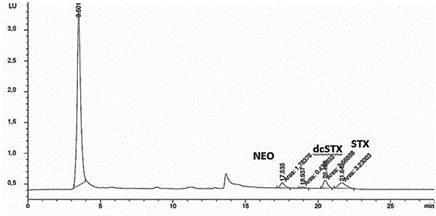
Figura 2 Cromatograma obtenido para las toxinas STXs al aplicar las condiciones cromatográficas establecidas por Rourke et al. (1). Orden de elución: NEO, dcSTX, STX.
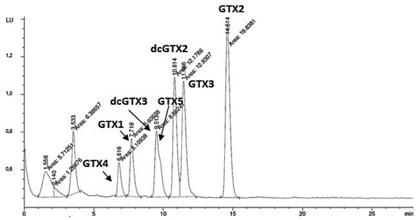
Figura 3 Cromatograma obtenido para las toxinas GTXs al aplicar las condiciones cromatográficas establecidas por Franco et al. (2).
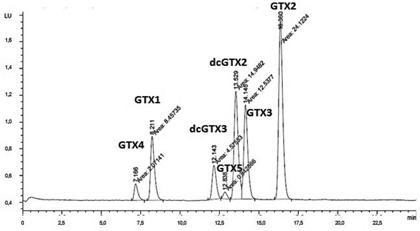
Figura 4 Cromatograma obtenido para las toxinas GTXs al aplicar las condiciones cromatográficas establecidas por Rourke et al. (1). Orden de elución: GTX4, GTX1, dcGTX3, GTX5, dcGTX2, GTX3, GTX2.
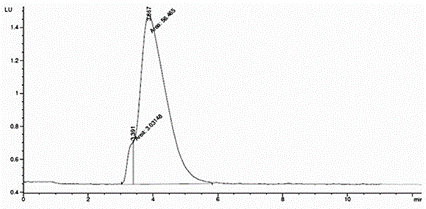
Figura 5 Cromatograma obtenido para las toxinas C1 y C2 al aplicar las condiciones cromatográficas establecidas por Franco et al. (2).
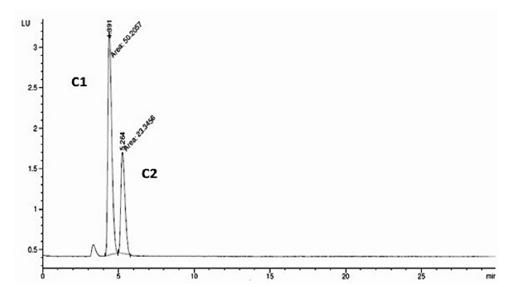
Figura 6 Cromatograma obtenido para las toxinas C1 y C2 al aplicar las condiciones cromatográficas establecidas por Rourke et al. (1). Orden de elución: C1, C2.
Con los resultados obtenidos en los cromatogramas, se consideró la metodología de Rourke et al. (1) como la idónea para el análisis de todos los grupos de toxinas en las muestras objeto de estudio.
Validación del método
A continuación se detallan los resultados obtenidos en la validación del método seleccionado, para el análisis de las toxinas evaluadas.
Linealidad y rango de trabajo
Se definió el rango de trabajo como el intervalo de las concentraciones en las que se aplicó el método. Se evaluó también la linealidad del método para cada toxina, es decir el rango de concentraciones del analito en el que la respuesta del sistema de medición, en este caso el área del pico cromatográfico, es una función lineal de la concentración.
De acuerdo con las curvas de calibración se estableció el ámbito lineal para cada analito, tanto en disolución como en matriz, mismo que se indica en el cuadro 2.
Cuadro 2 Determinación del rango de trabajo del método empleado para la determinación de toxinas PSP.
| Parámetro | Neo | dcSTX | STX | GTX4 |
| Rango lineal (µmol/L) | (0,1181-2,3616) | (0,0585 - 1,1700) | (0,0597 - 1,1934) | (0,0831 - 1,3298) |
| Rango lineal (µg/100 g) | (8,1-162,2) | (4,2-84,0) | (4,8-96,8) | (7,4-119,3) |
| Parámetro | GTX1 | dcGTX3 | GTX5 | dcGTX2 |
| Rango lineal (µmol/L) | (0,2548 - 4,0770) | (0,0413 - 0,6615) | (0,1371 - 2,1932) | (0,1408 - 2,2523) |
| Rango lineal (µg/100 g) | (22,8-365,6) | (3,2 - 50,8) | (11,3-181,4) | (10,8-172,9) |
| Parámetro | GTX3 | GTX2 | C1 | C2 |
| Rango lineal (µmol/L) | (0,0610 - 0,9765) | (0,1606 - 2,5695) | (0,2041 - 4,0824) | (0,0610 - 1,2204) |
| Rango lineal (µg/100 g) | (5,3-84,2) | (13,8-221,5) | (21,2-423,1) | (6,3-126,4) |
La similitud de las curvas de calibración preparadas para cada toxina, se evaluó mediante las pruebas de Tuckey y Fisher a un 95% de confianza. Los resultados indican que no existe diferencia significativa entre las curvas preparadas para un mismo analito, por lo que se graficó la curva promedio para cada uno. Para cada curva se determinó el coeficiente de correlación, el coeficiente de determinación y la ecuación de la recta de calibrado, tal como se observa en el cuadro 3. Para el caso específico de las toxinas Cs, la calibración fue externa (en disolución), mientras que para las GTXs y STXs, la calibración se realizó sobre matriz.
Cuadro 3 Evaluación de la linealidad del método empleado para la determinación de toxinas PSP.
| Parámetro | Neo | dcSTX | STX | GTX4 | GTX1 | dcGTX3 |
| Coeficiente de correlación (R) | 1,000 | 0,999 | 0,999 | 0,995 | 0,999 | 0,996 |
| Coeficiente de determinación (R2) | 1,000 | 1,000 | 1,000 | 0,990 | 0,999 | 0,992 |
| Parámetro | GTX5 | dcGTX2 | GTX3 | GTX2 | C1 | C2 |
| Coeficiente de correlación (R) | 1,000 | 0,995 | 1,000 | 1,000 | 1,000 | 1,000 |
| Coeficiente de determinación (R2) | 1,000 | 0,989 | 0,999 | 1,000 | 1,000 | 1,000 |
Según los datos mostrados en el cuadro 3, tanto el coeficiente de correlación, como el de determinación cumplen con los criterios establecidos por el procedimiento de aseguramiento de la calidad de la Unidad RECAA, según el cual R ≥ 0,99 y R2 ≥ 0,98 (21).
El rango lineal del método para la determinación de las toxinas PSP, expresado como µg STX diHCl equivalentes/100 g, es de 53,7 - 926,6. El mismo se estableció a partir del primer y último punto de la curva de calibración de cada analito. El rango lineal se considera aceptable, ya que el límite máximo permitido de 80 µg STX diHCl equivalentes/100 g está contemplado en el mismo (19).
Sensibilidad
La sensibilidad de calibración se determinó con la pendiente de la ecuación de la curva de calibración promedio de cada analito, los valores obtenidos se indican en el cuadro 4.
Cuadro 4 Determinación de la sensibilidad del método empleado para la determinación de toxinas PSP.
| Parámetro | Neo | dcSTX | STX | GTX4 | GTX1 | dcGTX3 |
| Sensibilidad LU*(µmol/L)-1 | 5,0462 | 10,296 | 15,982 | 4,3205 | 5,925 | 29,187 |
| Parámetro | GTX5 | dcGTX2 | GTX3 | GTX2 | C1 | C2 |
| Sensibilidad LU*(µmol/L)-1 | 5,804 | 21,895 | 60,322 | 24,277 | 10,188 | 20,944 |
Según el cuadro 4, la toxina que presentó un valor de pendiente menor, y por tanto una mayor sensibilidad fue la GTX4, mientras que la GTX3 mostró una sensibilidad menor comparada con los otros analitos.
Exactitud
Para conocer la exactitud del método, se realizó la evaluación de la veracidad y de la precisión.
En el cuadro 5 se muestra el nivel de concentración al que se realizó la fortificación, el porcentaje de recuperación obtenido para cada toxina, así como el criterio de aceptación establecido en la normativa para evaluar la veracidad. La C2 presentó el porcentaje de recuperación más bajo de las toxinas evaluadas (58%), lo cual se puede atribuir a una extracción incompleta del analito de la matriz en estudio. La GTX2 mostró el porcentaje de recuperación más alto (115%), lo cual se justifica en términos de la distribución normal que pueden seguir los datos de la concentración del analito en la muestra.
Según los datos indicados, para todas las toxinas PSP se cumple con el criterio de aceptación para el porcentaje de recuperación, establecido según el CODEX STAN 292-2008 de (50 - 130)% (19). Este rango de aceptación es el producto del consenso de expertos del CCMAS (Comité de Métodos de Análisis y Muestreo) del Codex Alimentarius, los cuales usan como base científica los datos de validación de los laboratorios de referencia de los países miembros.
Cuadro 5 Determinación de la veracidad del método empleado para la determinación de toxinas
| Parámetro | Neo | dcSTX | STX | GTX4 | GTX1 | dcGTX3 |
| Sensibilidad LU*(µmol/L)-1 | 5,0462 | 10,296 | 15,982 | 4,3205 | 5,925 | 29,187 |
| Parámetro | GTX5 | dcGTX2 | GTX3 | GTX2 | C1 | C2 |
| Sensibilidad LU*(µmol/L)-1 | 5,804 | 21,895 | 60,322 | 24,277 | 10,188 | 20,944 |
PSP.
Los valores obtenidos en la evaluación de la precisión del método se pueden observar en el cuadro 6 .
En todos los casos, se cumple el criterio de aceptación para los analitos, debido a que los valores de CV (%) obtenidos en las pruebas de repetibilidad son inferiores a los establecidos por CODEX STAN 292-2008 (44% para Neo, dcSTX, STX, GTX4 y GTX1 y 38% para dcGTX3, GTX5, dcGTX2, GTX3, GTX2, C1 y C2) (19). La evaluación de la repetibilidad instrumental y de la metodología, indica que el método para la determinación de toxinas PSP por cromatografía líquida es preciso.
Cuadro 6 Evaluación de la precisión del método empleado para la determinación de toxinas PSP.
| Parámetro | Neo | dcSTX | STX | GTX4 | GTX1 | dcGTX3 |
| CV (%) Repetibilidad Instrumental | 3 | 2 | 2 | 17 | 6 | 17 |
| CV (%) Repetibilidad de la metodología | 6 | 3 | 3 | 16 | 7 | 4 |
| Parámetro | GTX5 | dcGTX2 | GTX3 | GTX2 | C1 | C2 |
| CV (%) Repetibilidad Instrumental | 15 | 13 | 16 | 5 | 1 | 1 |
| CV (%) Repetibilidad de la metodología | 8 | 3 | 3 | 3 | 4 | 2 |
Límites de detección y cuantificación
En el cuadro 7 se muestran los valores obtenidos para los límites de detección y de cuantificación calculados, utilizando la metodología IUPAC (22).
Cuadro 7 Determinación de los límites de detección y cuantificación del método empleado para la determinación de toxinas PSP.
| Parámetro | Neo | dcSTX | STX | GTX4 | GTX1 | dcGTX3 |
| Límite de detección (µg/100 g) | 1,3 | 2,1 | 2,5 | 7,0 | 1,4 | 1,1 |
| Límite de cuantificación (µg/100 g) | 5,2 | 5,0 | 6,1 | 9,4 | 4,6 | 1,6 |
| Parámetro | GTX5 | dcGTX2 | GTX3 | GTX2 | C1 | C2 |
| Límite de detección (µg/100 g) | 8,5 | 1,2 | 2,1 | 7,4 | 2,8 | 1,8 |
| Límite de cuantificación (µg/100 g) | 12,1 | 1,8 | 2,4 | 12,6 | 3,9 | 3,9 |
Con los datos obtenidos en el cuadro 8 se define una concentración de STX menor o igual a 2,5 µg/100 g como “No detectado”. Concentraciones mayores a 2,5 µg/100 g y menores a 6,1 µg/100 g se reportan como “No cuantificable”.
Al evaluar la veracidad en los niveles del límite de detección y cuantificación, se obtuvieron los valores de porcentaje de recuperación detallados en el cuadro 8.
Cuadro 8 Evaluación de la veracidad en el límite de cuantificación.
| Parámetro | Neo | dcSTX | STX | GTX4 | GTX1 | dcGTX3 |
| Concentración cercana al límite de detección (µg/100 g) | 1,8 | 2,1 | 2,8 | 2,3 | 7,1 | 1,1 |
| Concentración cercana al límite de cuantificación (µg/100 g) | 5,6 | 5,0 | 6,3 | 4,6 | 14,0 | 1,6 |
| Porcentaje de Recuperación en el LC | 95 | 132 | 74 | 121 | 76 | 174 |
| Parámetro | GTX5 | dcGTX2 | GTX3 | GTX2 | C1 | C2 |
| Concentración cercana al límite de detección (µg/100 g) | 8,5 | 3,9 | 2,86 | 7,52 | 6,06 | 1,81 |
| Concentración cercana al límite de cuantificación (µg/100 g) | 12,1 | 5,4 | 4,8 | 12,6 | 13,3 | 4,0 |
| Porcentaje de Recuperación en el LC | 112 | 131 | 118 | 103 | 72 | 82 |
Según el cuadro 8, la evaluación del límite de detección permitió comprobar que en todos los casos, se logra identificar el analito en cuestión. Al evaluar la veracidad en el límite de cuantificación, se presentaron porcentajes de recuperación que incumplen los criterios de veracidad de 50% a 130%, establecidos en la normativa CODEX STAN 292-2008 (19), por ejemplo: 132% para la dcSTX, 174% para la dcGTX3 y 131% para la dcGTX2. Por lo anterior se recomienda que estos sean reevaluados cuando el método se establezca como rutina en el laboratorio. No obstante, la contribución de todos los límites de cuantificación, al igual que los de detección, se emplearon para establecer los parámetros de desempeño del método.
El límite de detección y límite de cuantificación expresados como µg STX diHCl equivalentes/100 g se determinó en 19,8 y 40,7 respectivamente. Estos valores se consideran aceptables, ya que según el Codex Alimentarius (CX/RVDF 03/10) (23) para las sustancias que tienen límite máximo de residuos (LMR), el límite de cuantificación (LC) debe ser al menos la mitad del LMR. En este caso el límite máximo es de 80 ug STX diHCl equivalentes /100 g de carne de molusco (19). La evidencia obtenida en la determinación de los parámetros de desempeño, permitió concluir que el método para la determinación de toxinas paralizantes por HPLC/FLD con oxidación post columna, cumple los parámetros de validación requeridos para su aplicación.
Conclusiones
Se logró la separación, identificación y cuantificación de las toxinas PSP al emplear el método cromatográfico establecido por Rourke et al. (1). El método de extracción de las toxinas PSP en moluscos bivalvos aplicado en este estudio permitió cumplir con los criterios de sensibilidad y exactitud indicados en la normativa vigente. La evidencia obtenida en la evaluación de los parámetros de desempeño: linealidad, ámbito de trabajo, sensibilidad, especificidad, selectividad, exactitud (veracidad y precisión), límites de detección y límite de cuantificación, permitieron concluir que el método para la determinación de toxinas paralizantes por HPLC/ FLD con oxidación post columna, es apto para su aplicación. El contar con un método analítico cuantitativo para la determinación de toxinas PSP, permitirá al SENASA (institución responsable de velar por la inocuidad de los alimentos de origen animal), específicamente en la dirección del LANASEVE (laboratorio oficial encargado de ejecutar los análisis de los contaminantes presentes en los alimentos), obtener resultados cuantitativos confiables, los cuales se pueden comparar con la normativa de referencia internacional: Codex Alimentarius y Unión Europea.
Agradecimientos
Al Laboratorio Nacional de Servicios Veterinarios (LANASEVE) y al Servicio Nacional de Salud Animal (SENASA) por todo el soporte instrumental. Al Instituto Costarricense de Pesca y Acuicultura (INCOPESCA) por la cooperación en el muestreo y envío del material para la elaboración del presente trabajo.

